您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2023-02-11 05:58
药厂无菌制剂车间使用的自动配料系统,主要由浓配罐、稀配罐、无菌储罐、缓冲罐、管道以及 PLC 控制系统组成,用于无菌药品的溶解、配制。配料系统是无菌制剂生产过程中最关键的工序,为下一工序提供符合要求的中间体,是产品质量的有效保证。文章通过对自动配料系统性能测试相关技术进行研究,测试设备称重、搅拌均匀度、在线清洗(CIP)以及在线灭菌(SIP)等功能,并进行称重、核黄素和生物挑战性等检查,考察其性能测试方法的可靠性和有效性,不断完善系统自身的性能测试方法,提高无菌药品生产的质量,保障用药安全。
药液的配制是指灌装前将原辅料和溶剂混合的过程,无菌制剂的产品分为最终灭菌产品和非最终灭菌产品,而非最终灭菌产品作为高风险的品种,其配制过程,则需要尽可能的采取措施,防止污染和交叉污染,采用具备自动清洗和自动灭菌功能的自动化配料系统进行无菌药品的配制,可以有效降低微生物污染的风险,符合 GMP 的要求。
无菌制剂车间的自动配料系统,位于车间的 C 级洁净区,是无菌药品生产过程中最关键工序配料所使用的设备,为下一工序提供符合质量要求的中间体。按照无菌药品工艺规程完成该配料过程,是产品质量有保证的前提,而自动配料系统安装完成后,需对该系统进行设计、安装、运行以及性能等测试,性能测试则作为整个过程中的重中之重。本文侧重研究自动配料系统的性能测试方面,设计了数项测试技术,验证其压力保持、升降温速度、称重功能、搅拌功能、在线清洗(CIP)以及在线灭菌(SIP)等功能,通过测试数据结果证明该自动配料系统能够按照既定工艺完成药品的配制工序,提高了无菌药品中间体的质量。
Part1 自动配料系统描述及工作原理
自动配料系统主要用于药品生产过程中产品的配制过程,主要由浓配罐、稀配罐、无菌储罐、缓冲罐、管道以及PLC 控制系统组成,整套系统具有精密采水定容、系统在线CIP 和 SIP、吹干保压等功能,系统的排水口具有隔断排水装置,防止灭菌后冷却形成的负压倒吸,稀配罐按工艺进行定容,经过药液过滤器过滤暂存罐,可以在暂存罐对其进行无菌取样。在取得灌装机供液信号后,将药液经除菌过滤器过滤后压入缓冲罐,供灌装机灌装药液。
以上自动配料系统的配料罐均采用不锈钢 SUS316L,罐体设有夹套,可加热、冷却或保温。内表面采用镜面抛光,无卫生死角,全封闭的设计确保物料始终处在隔离状态下。封头均经旋压加工,搅拌装置采用卫生级机械密封,接口采用 ISO 标准快装卡箍式,均符合 GMP 标准。设备配有 CIP清洗喷头、视镜、法兰式快开人孔等装置,溶解罐、精配罐和暂存罐均配有无菌空气呼吸器、称重模块。自动配料系统的工作原理,首先是在浓配罐进行药液配制,然后在稀配罐按药品工艺进行定容,药液经过除菌过滤器过滤至无菌储罐(可在无菌储罐对其进行无菌取样),在取得灌装机供液信号后,最终将药液经除菌过滤后压入缓冲罐,供灌装机灌装药液,其稀配罐和无菌储罐均配置无菌空气呼吸器和称重模块,并配有 CIP 清洗喷头,可通过视镜进行观察,其法兰式快开人孔装置可提供物料的进入。
Part2 性能测试部分
1 材料与设备
材料与试剂:注射用水、嗜热脂肪杆菌芽孢生物指示剂、核黄素。仪器与设备:电子秤、黑光灯、温度验证探头、可见分光光度计、生化培养箱。
2 测试方法
压力保持效果测试
压力保持效果测试,是为了确认自动化配料系统在运行或者停止的状态下,对其进行加压,使系统内部和外部环境完全隔离,测试其压力保持的效果,看是否能杜绝交叉污染和安全隐患。
(1)测试步骤:①手动对配料系统进行加压至 0.15 MPa~0.20 MPa(采用压缩空气加压);②加压完成后,确认所有排污阀门、过滤器阀门等处于关闭状态;③记录各阀门压力传感器上的初始压力;④待时间经过 24 h 以上,记录各个阀门上压力表读数。
(2)判定标准:保压应能持续 24 h 以上,压力应能持续保持正压,且压力变化值应不能超过 0.04 MPa。
(3)试验结果:压力保持测试结果如表 1 所示。
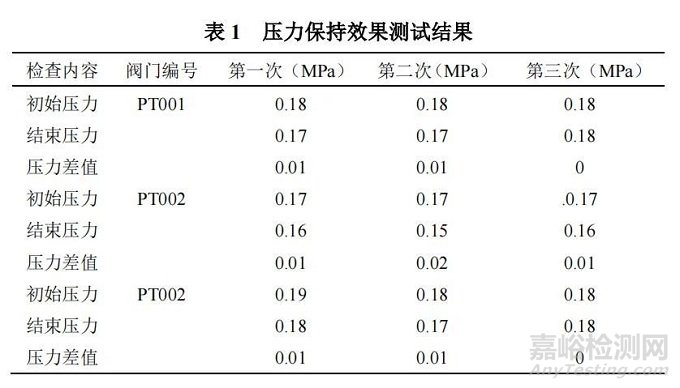
由表 1 可以看出,罐体经过三次 24 h 的压力保持测试,压力变化值均未超过 0.04 MPa 的标准值,说明该配料系统在规定时限内能保持正压,能有效防止外界污染气体进入该系统造成污染的后果,该测试方法有效。
浓配罐升降温速度及恒温效果测试
在系统自动模式下运行过程中,通过浓配罐罐体夹套升降温测试,考察其温度是否在要求范围内,检查其升降温以及恒温效果是否符合要求。
(1)测试步骤。①浓配罐采集注射用水 300 L,模拟工艺溶解步骤,搅拌 80 min;②对罐体夹套通入蒸汽,记录初始温度及罐体温度达到 80℃的时间;③罐体维持 80℃,记录通入蒸汽保持罐体 80℃的时间;④对罐体通入冷冻水,记录罐体从 80℃下降到 60℃的时间。
(2)判定标准。罐体应能保持工艺所需温度(80℃)30 min;罐体升温、降温所需时间应小于 30 min。
(3)试验结果。升降温速度及恒温效果检查结果如表 1 所示。
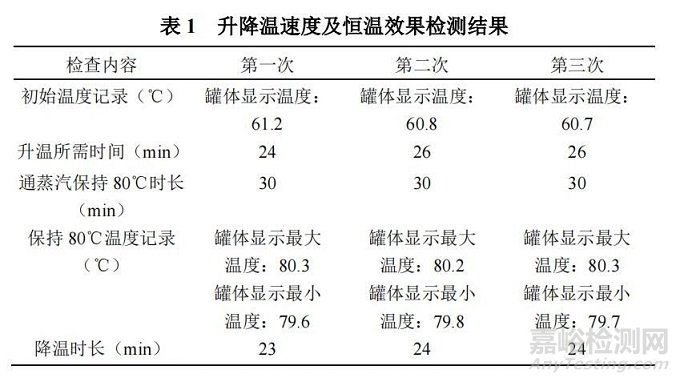
由表 1 可以看出,罐体的升降温速度均在合格范围内,且罐体能保持工艺所需温度,说明罐体升降温及恒温性能测试符合要求。
称重功能测试
通过采用标准砝码对稀配罐进行称量准确度测试,考察其称重偏差是否在合格范围内。
(1)测试步骤:①对稀配罐称重模块数值调零;②在稀配罐内逐个增加标准砝码(每个 20 kg)至 120 kg,并从显示屏读出每次称重模块累计重量;③取读数与砝码累加重量进行比较,计算称重偏差。
(2)判定标准:称重偏差应≤±0.5%。
(3)测试结果:称重功能测试结果如表 2 所示。
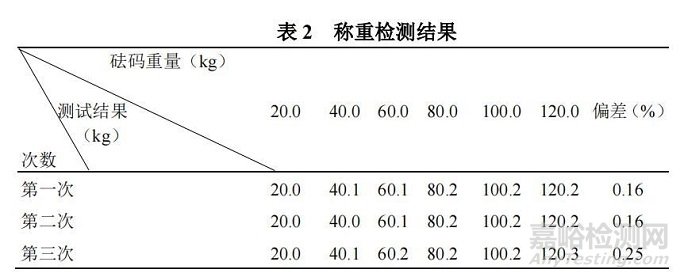
通过三次测试数据表明,稀配罐称重测试数值与标准砝码之间偏差均在合格标准范围内,测试方法有效、准确。
搅拌均匀度测试
药液配制过程中,由于药液有粘稠性,不同的搅拌速度下其溶解过后均匀度也不尽相同,为了考察浓配罐的搅拌均匀度,可以设置不同的转速,如 75%、100%、125%,以相似性质的物料代替药液加入至浓配罐中,待搅拌均匀后,可在罐内不同部位取样检测,通过考察搅拌均匀性,来检测该搅拌系统是否达到搅拌均匀的要求。
(1)检测方法如下。
①于浓配罐内加入 3 kg 牛血清蛋白,并加入 300 L 注射用水稀释成每 1 mL 含 10 mg 的溶液。
②分别设置转速 45 r/min、60 r/min 和 75 r/min 进行搅拌,各搅拌 20 min。
③搅拌结束后,分别于罐体内部的上、中、下层以及每层的左、中、右各取一个点的样品进行蛋白质含量检查,并计算蛋白质含量 RSD 值。
(2)判定标准。
在罐体不同搅拌速度下,不同位置各个取样点蛋白质含量 RSD 应≤2.0%。
(3)试验结果。
蛋白质含量测试结果如表 3 所示。
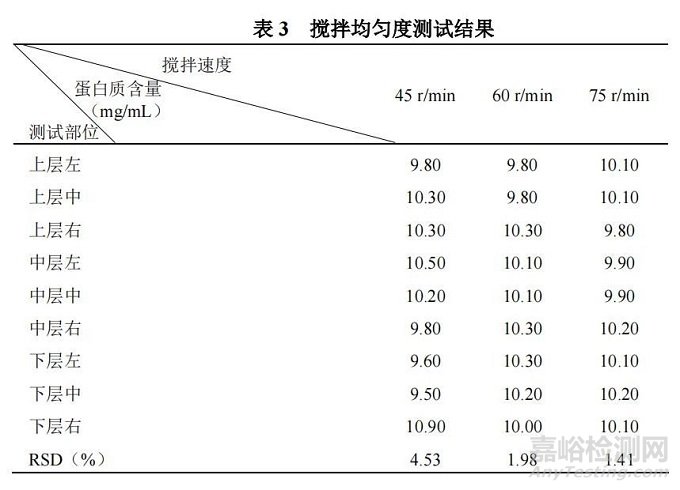
通过测试数据表明,45 r/min 转速下,蛋白质含量 RSD为 4.53%,结果不符合测试要求,但 60 r/min 和 75 r/min 转速下,含量 RSD 分别为 1.98%和 1.41%,结果符合要求,说明设备可采用 60 r/min 的速度进行搅拌,此搅拌速度下药液可充分搅匀。
在线清洗(CIP)检测
通过在罐体内部喷洒核黄素,启动自动配料系统进行喷淋冲洗,采用黑光灯(365 nm)进行观察是否有明显黄色荧光,来考察自动配料系统经过自动清洁程序后,是否清洁完全,无异物残留。
(1)检测方法。
①取核黄素适量,配成 0.2 g/L 的溶液,分别喷洒于浓配罐、稀配罐、过滤器、缓冲罐内表面,并确保罐体内表面可被均匀喷洒;
②用黑光灯(365 nm)进行检查,确定有明显的黄色荧光;
③待罐体干燥 1h 后,启动自动配料系统在线清洗(CIP)程序,清洗后,用黑光灯进行检查。
(2)判定标准。
按在线清洁程序进行清洁后,设备内部在黑光灯下应无荧光。
(3)试验结果。
在线清洗(CIP)测试结果如表 4 所示。

通过检测数据表明,经过自动配料系统在线清洗过后,均无观察到荧光存在,说明系统清洁完全,可按既定清洁程序进行清洗罐体。
在线灭菌(SIP)检测
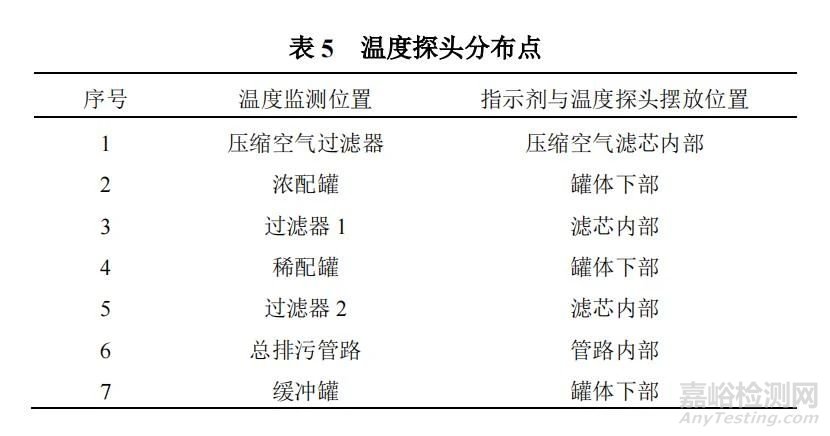
由于无菌产品的特殊性,自动配料系统在经过配液、清洗过后,需要对系统进行在线灭菌(SIP)操作,以去除微生物,防止微生物的滋生影响药品质量。设置系统灭菌温度121℃,灭菌 30 min,采用温度验证探头对自动配料系统灭菌程序进行监测,并进行生物挑战性试验,可确保系统经过在线灭菌程序后,证实充分杀死细菌,保障药品生产过程。
(1)检测方法。
①采用无线温度探头与有线温度探头结合的方式,将探头放置于罐体及管路不同位置,且温度探头尽可能安放在低温点。
②取嗜热脂肪杆菌芽孢生物指示剂放置于温度探头位置处进行生物挑战性试验。
③启动在线灭菌程序,并对温度进行监测。
(2)判定标准。
①温度探头温度均≥121℃。
②最低 F0 值应≥12。
③生物指示剂阳性对照试验应呈正反应。
④灭菌后的生物指示剂经过培养后应呈负反应(即无菌、培养基无颜色变化)。
(3)试验结果。
在线灭菌杀灭细菌测试结果如表 6 所示。
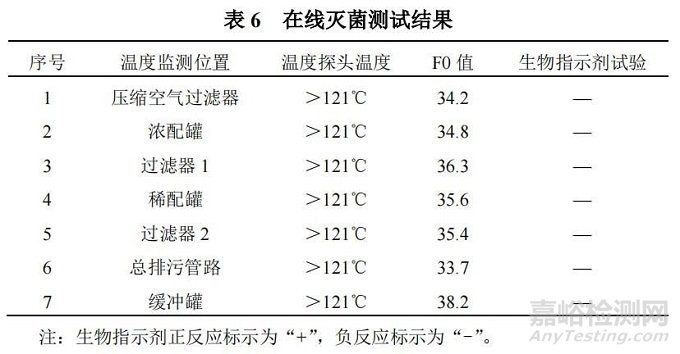
通过测试数据表明,系统在线灭菌过程后,各检测点温度均大于 121℃,灭菌 F0 值均大于 12,且生物挑战性试验结果均为负反应,说明系统性能检测合格,符合验收要求,通过该测试项目,可以准确反映系统灭菌效果,为下一批次产品的生产提供无菌保障。
Part3 结论
通过对自动配料系统性能测试技术的研究,其测试结果完全符合要求,证明了该测试技术的可靠性和有效性,并由此说明该性能测试方案是合乎日常生产实际的。然而,系统的性能测试方法通常不止上述这几项测试方法,技术人员还可以从中开发更多测试项目,完善自动配料系统的性能测试方法,从而发现系统运行过程中存在的问题,减少由于系统自身问题存在的缺陷,保证药品生产的质量。
参考文献
[1] 国家食品药品监督管理局药品认证管理中心. 药品 GMP指南——无菌药品[M]. 北京: 中国医药科技出版社,2011.
[2] 张志峰. 无菌全自动化配液系统在线灭菌解决方案[J]. 中国制药信息,2015(3): 14-17.
[3] 马义岭,郭永学,王云宝,等.制药设备与工艺验证[M].北京: 化学工业出版社,2019.

来源:制药工艺与装备


