您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-09-21 14:10
处方前研究在剂型与制剂的设计和改进工作中已逐步成为常规化的研究项目,为进一步设计剂型及优化制剂的处方工艺提供依据。
处方前研究工作的主要内容是通过查阅国内外文献资料或进行实验研究,获得剂型与制剂设计所需的各种资料,包括药物的物理性状、熔点、粒子大小、溶解度、溶出速度、晶型、pKa、分配系数、表面特性等药物固有的物理化学性质,药物的稳定性,药物与辅料的相容性,药物的药理、毒副作用与刺激性,以及药物的体内吸收、分布、代谢和排泄规律等。
1 药物的理化性质测定
粒子大小与分布
药物的某些理化性质(如药物的溶解度、溶出速度、稳定性等)以及制剂操作与质量(如粉体流动性、含量均匀度、生物利用度等)常受粒径与粒径分布的影响。
多数固体制剂(如散剂、颗粒剂、片剂、胶囊剂等)需要进行粒子加工以改善粉体性质来满足产品质量和粉体操作的需求。颗粒大小显著影响某些药物的口服吸收,如灰黄霉素、螺内酯等。因此,及早了解药物粒子大小对制剂处方和产品有效性的影响十分重要。
根据粒径不同,测定药物粒径及粒径分布可选用筛分法(药典筛)、沉降法(库尔特计数器)显微镜法(光学显微镜、扫描电镜、透射电镜)等。
溶解度
药物的溶解性是药物剂型与制剂设计时需要考虑的首要因素之一,药物无论通过何种途径给药,都必须具有一定的水溶性才能被机体吸收并产生治疗作用。
溶解度差的药物(水中溶解度<10mg/ml)往往表现为不完全或不稳定的吸收。如果药物的溶解度低于期望值,则需要考虑采用适宜的方法增加其溶解度。
药物溶解度的测定常采用摇瓶法。一般是将过量药物置于欲测定的溶剂内,在一定温度下振摇,测定达到平衡后的药物浓度即为其溶解度。通常需振摇60~72 小时才能达到平衡。
在处方前研究中,常需要测定药物在多种溶剂中的溶解度,如水、0.9%NaCl溶液、稀盐酸溶液(0.1mol/L HCI)、稀碱溶液(0.1mol/L NaOH)、pH6.8磷酸盐缓冲液和乙醇、甲醇等某些特定溶剂。这种测定方法测得的溶解度又称为平衡溶解度(equilibrium solubility)或表观溶解度(apparent solubility)。
溶出速度
对于难溶性药物及其口服剂型,如片剂、胶囊剂、混悬剂,以及混悬剂肌内注射给药,其药物在吸收部位体液中的溶出是药物吸收的限速步骤。当溶出是限速过程时,任何影响溶出的因素也将影响药物吸收。实际工作中,通常采用体外溶出度测定法测定药物的溶出速率来预测药物在机体内的溶出和释放。
目前,《美国药典》(USP35)收载有多种溶出度检测方法,包括篮法、桨法、往复筒法、流池法、桨碟法、转筒法、往复支架法等。
《中国药典》2020年版四部通则规定有3种溶出度测定方法(0931),包括第一法(篮法)、第二法(桨法)、第三法(小杯法)、第四法(桨碟法)、第五法(转筒法)、第六法(流池法)、第七法(往复筒法)。根据不同的研究和应用目的可选择不同的方法和仪器。
溶出度测定的应用日益广泛,包括药品研发阶段的处方筛选、制剂常规质量控制、评价药品有效期内的质量、预测不同服药条件下( 如饭前、饭后 服药) 药物释放对药物有效性的影响、评价药品的生物等效性、评价上市后药品组分改变和生产方法改变后的质量等。总之,溶出度测定在控制药品质量、预测药品有效性等方面发挥着重要作用。
分配系数
分配系数(partition coefficient,P)是指药物在两个不相混溶的溶剂中溶解并达到平衡时浓度的比值。油/水分配系数(例如正辛醇/水、三氯甲烷/水)是表示药物分配在油相和水相中的比例。
药物在体内的溶解、吸收、分布、转运与药物的水溶性和脂溶性有关,即与药物油/水分配系数有关。一般而言,油/水分配系数较大的药物更容易穿透细胞膜转运和吸收。
由于正辛醇的溶解度参数与生物膜溶解度参数接近,为21.07(J/cm³)二分之一,因此,为了更准确地预测药物在体内的转运,在处方设计中常使用正辛醇/水分配系数,并且常采用摇瓶法测定。
如用正辛醇饱和的水和用水饱和的正辛醇按照1:1混合后,加入标准量的药物,在恒定温度下振摇至平衡,测定两相中的药物浓度C油和C水,求算比值C油/C水即得。
如果药物在两相中都是以单体存在,则分配系数为药物在两相中的溶解度之比,只要测定两个溶剂中药物的溶解度即可求得分配系数。
解离常数与pKa
药物解离程度对药物的处方以及其动力学参数均具有重要影响,是值得关注的理化特性之一。通常情况下,药物的解离程度极大地取决于含药介质的pH,在处方工艺研究中常常采用调整介质pH的办法,使得药物在一定程度上离子化以满足所需的药物溶解度和制剂稳定性。
药物解离程度对药物的吸收、分布、消除也有重要影响。一般而言,解离型药物较难通过生物膜而被吸收,而非解离型药物往往可有效地通过类脂性的生物膜。溶液pH、药物的pK,以及药物解离状态的关系可以用 Henderson-Hasselbalch 方程表示。
解离常数或pKa通常可以由电位滴定法测定。根据 Henderson-Hasselbalch方程,如果已知Ca和pKa,则可预测任何pH条件下药物的溶解度(非解离型和解离型溶解度之和)。利用解离常数或pKa,可以预测在一定pH时混合是否沉淀,同时还有助于选择药物的合适盐。
药物的多晶性
晶型(crystalline forms,polymorphs) 是指晶态物质晶格内分子的排列形式。药物常存在有一种以上的晶型,称为多晶型,又称同质多晶现象(polymorphism)。
许多药物具有多晶型,同种药物由于晶型结构不同,某些物理性质如密度、溶解度、溶出速度等不同,可能会影响药物的体内溶出、吸收,进而可能在一定程度上影响药物的临床疗效和安全性,尤其是一些难溶性药物的口服固体或半固体剂型,晶型的影响更大。除了多晶型外,药物分子也可能以无定型存在,无定型总是比相应的结晶型具有更大的溶解性。
药物的多晶型评价是一项重要的处方前研究内容。难溶性的固体药物如需制成固体口服制剂,应对原料药的晶型进行研究,因为制备工艺等均可能引起药物晶型的转变。
一个新的化合物,首先应该研究是否存在多晶型,有多少种晶型,是否存在无定型,并研究不同晶型的理化性质差异(如溶解度、稳定性)等,确定目标晶型,并通过药理毒理及临床试验,确定所选晶型的优劣。
如果对药物的多晶型研究不得当,有可能引起制剂品种的一系列问题,如结晶析出、晶型转变、稳定性差、生物利用度低等。如注射用醋酸可的松混悬液,若用错了晶型,久置会发生结块现象。对于有晶型的药物,在稳定性考察试验中应设置晶型考察指标,以确定适宜的贮存条件,确保晶型稳定。
研究药物多晶型时,常使用的方法包括热台显微镜法、热分析法、红外光谱法和X射线衍射法等。
药物的吸湿性
吸湿性(hygroscopicity)是指固体表面能从周围环境空气中吸附水分的现象。药物粉末由于比表面积大,大多具有不同程度的吸湿性,在湿度较大的空气中容易不同程度地吸附一些水分,导致润湿、流动性下降、结块、液化等,甚至出现变色分解等而降低药物的稳定性。药物吸附水分的速度和程度取决于药物的理化性质和环境的相对湿度(relathumidity,RH)。
药物的吸湿性常用吸湿平衡曲线表示,即在不同湿度下测定药物的平衡吸湿量,再以平衡吸湿量对相对湿度作图即得。水溶性药物与水不溶性药物的吸湿平衡曲线有显著差异。CRH 是水溶性药物粉末的特征参数,水溶性药物在环境相对湿度低于 CRH下时几乎不吸湿,而当相对湿度增至CRH,吸湿量急剧增加。水不溶性药物随空气中相对湿度的增加缓缓吸湿,无临界值。水不溶性药物混合物的吸湿性具有加和性。
测定药物的CRH时,可将药物粉末置于不同已知相对湿度的环境中(如贮于恒温恒湿箱或具有饱和盐溶液的干燥器中)进行试验,当吸湿达平衡时称重计算吸水量(增重),以平衡吸湿量对各个相对湿度作图得吸湿平衡曲线,曲线斜率急剧变化处的相对湿度即为该样品的CRH。
药物的稳定性
热、光、氧气、水分、pH以及辅料等对药物的稳定性都可能产生重大影响。因此,处方设计前的一个重要工作就是对影响药物稳定性的因素进行考察。通过对药物本身稳定性的研究,可对处方组成、制备工艺、辅料和稳定性附加剂的选用和合适的包装设计起重要的指导作用。
药物的稳定性试验是研究热、氧气、水分及光线对药物稳定性的影响,同时也可用来确定合适的包装材料、贮存技术和方法等。
2 生物药剂特征与药学动力参数
药物的吸收、分布与消除
给药后,药物在体内必须穿透生物膜,才能被吸收进人血液循环系统。药物的理化性质对其吸收有很大影响。现代的处方前研究包括了药物分子透过生物膜的早期评价。
由处方前药物理化性质研究获得的数据尤其是pKa、油水分配系数、溶解度和溶出速度,为研究药物吸收的可能性提供了依据。
体外小肠实验技术常被用于评价药物的吸收特性。
药物吸收后,经血液循环分布到全身各器官、组织中,同时通过代谢和排泄从体内消除。由于药物分布与消除速度决定了血液和作用部位药物的浓度,从而决定了给药的频率,所以在确定剂型和处方设计前应充分了解药物的分布、代谢和消除等特点。
不同的剂型、用药部位及给药途径,可以影响一种药物在体内的吸收,分布、代谢及排泄过程,从而产生不同的吸收速度、起效时间、达峰时间和作用持续时间。药物不同给药剂型、不同给药途径的体内转运过程及对药物吸收的影响见下图。
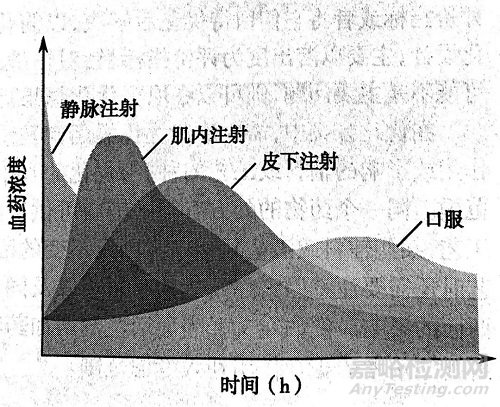
药物的体内动力学参数
在处方前研究工作中,应通过查阅文献,充分了解药物本身的体内动力学性质和参数,以便设计合适的给药途径和剂型。
肌内注射如药物的半衰期很短或很长,一般都不考虑制成缓释制剂。再如首过效应明显的药口服物,可选择非胃肠道给药或采用纳米给药系统口服给药以避免或减少首过效应对药物的影响。
药物的药理、药效、毒理特性
在制剂的设计过程中,还需要充分了解药物的药理、药效、毒理等特性,以确保在临床应用时能最大限度地发挥疗效,降低毒性。
毒理学特性
了解药物的毒理学性质对于制剂设计十分重要。制剂的设计应能提高药物的安全性,降低刺激性或毒副作用。如对于具有胃肠道不良反应的药物,一般不宜选择口 服给药的剂型;如果只是对胃具有刺激性,则可设计成肠道释药的剂型。
对具有皮肤剌激性的药物,应尽量避免皮肤给药。对毒性较大的药物,应选择可显著降低药物不良反应的缓控释剂型。对于治疗指数低的药物,宜设计成缓控释制剂,以减小峰谷波动,维持较稳定的血药浓度水平,降低毒副作用。
药理和药效学性质
在药物制剂设计的过程中,应当充分了解原料药物的作用机制和药效学性质,如药物的作用部位与靶点、治疗窗的范围、动物模型等,用以指导剂型和制剂的设计。
如治疗心绞痛药物的硝酸甘油通过舌下、透皮、口服等多种形式给药时,起效快慢与作用强度有很大的区别。如果是对心绞痛进行急救,宜选用舌下给药,使之迅速吸收。对于预防性质的长期给药,则使用透皮贴剂较为合适。

来源:Internet


