您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-09-08 14:59
遗传毒性杂质(GTIs)是一类在较低水平时也有可能直接引起DNA损伤,导致DNA突变,并可能引发癌症的DNA反应性物质。
2018年,华海药业缬沙坦原料药中致癌物质N-亚硝基二甲胺( NDMA)超标,导致数十批药品被召回,在国内医药圈引起轩然大波,国内正式开始关注遗传毒性杂质。CDE快速发布了针对遗传毒杂质研究的ICH M7翻译件,2020版中国药典也增加了遗传毒性杂质控制的指导原则,引导国内企业进行该类杂质的研究。本文通过对各类指导性文件的学习总结,结合自己的项目经验,对新药研发中遗传毒性杂质研究的一般流程进行论述,不足之处,还请各位老师指正。
一确定遗传毒性杂质类别
ICH M7根据杂质的研究情况将遗传毒性杂质分为以下5类
表1 遗传毒性杂质的分类及控制
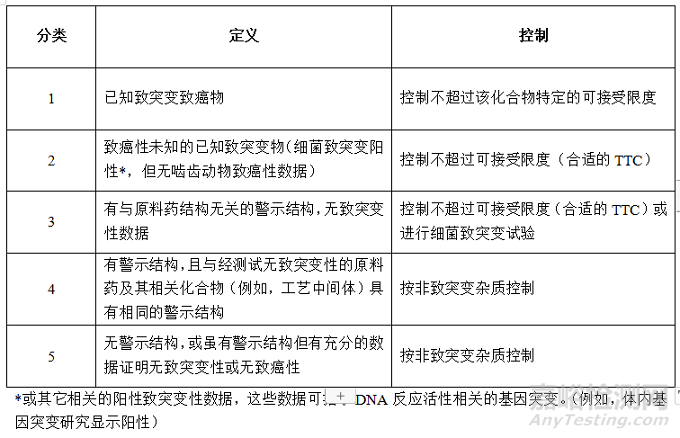
确定遗传杂质类别可按以下步骤进行:
1)列出需要研究的杂质列表,包括原料药和制剂生产和贮存期间可能产生的实际的和潜在的杂质。该项工作是之后所有工作的基础,需要合成、制剂及分析人员通力合作,确保杂质列表足够全面。
2)通过数据库和文献检索,查找各杂质的致突变性和致癌性数据,如有,则归为1类或2类。
3)对于没有致突变性致癌性数据的杂质,用(定量)构效关系[(Q)SAR]评估方法进行评估,预测细菌突变试验(Ames)的结果。应采用两种互补的(Q)SAR 预测方法。一种方法基于专家知识规则,另一种方法基于统计学。如果两个互补的(Q)SAR方法预测结果没有警示结构,则可将该杂质归为5类。如果某杂质(Q)SAR方法预测结果为阳性,或者两个(Q)SAR方法预测结果相矛盾,则分析其警示结构,如与原料药警示结构一致,则归为4类;不一致,暂时归为3类。
目前,(Q)SAR 预测的费用较高,但CDE对(Q)SAR 预测结果比较认可,建议尽量做一下,以免发补。如果想要降低成本,可参考遗传毒性杂质警示结构[1]文中所列的警示结构对杂质进行分析,只对含有警示结构的杂质进行(Q)SAR 预测。
4)对于3类杂质,如果制备成本尚可接受,可以进行该杂质的细菌突变试验(Ames),如果Ames试验结果为阴性,则该杂质归为5类;如果Ames试验结果为阳性,可以归为2类。Ames试验尽量在符合GLP的条件下进行,否则实验结果很可能被CDE质疑。
考虑到成本问题,如果进行到第4)步,杂质确定为2类,一般不再进行致癌性试验的研究,而是按照2类杂质进行控制。
二设定遗传毒性杂质的限度
对于1类杂质,由于限度已知,样品中杂质的限度可通过下式简单计算得出:
杂质限值=杂质可接受摄入量/药物每日最大用量
对于2、3类杂质,一般根据毒理学关注阈值(TTC)计算限度,即一个杂质的可接受摄入量为1.5μg/d;当有多个2、3类杂质时,其总限度需满足表2的限度要求,需注意1类杂质不计入总限度的计算中。
1.5μg/d 的摄人量是基于终生长期用药(>10年)的假设推导出来的,当实际用药时间较短时,其限度可适当放宽,表2为根据用药时间设定的限度。需要注意的是,对于间隔给药,治疗期是给药的总天数,而不是给药的时间间隔。
表2. 杂质的可接受摄入量
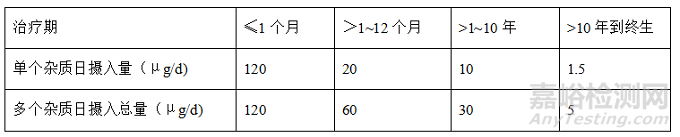
另外,毒性较强的黄曲霉素类、N-亚硝基化合物,以及烷基-氧化偶氮基化合物不适用TTC法,这些物质的阈值可以参考对应的结构类似物的阈值,如有多个类似物的阈值,优先选择限度较低的。
可以看出,以上1、2、3类杂质的限度多在ppm级,如果都在终点控制(成品质量标准中控制),限度较低,控制难度大。对于API中的工艺杂质,可以将控制位置前移,如在原料、起始物料或中间体质量标准中进行检测,设置一个较高的杂质限度,然后通过杂质的清除转化研究,证明该杂质可以被工艺有效去除,设定的较高的杂质限度是合理的,以此减轻分析工作压力。
对于4、5类杂质,按照一般杂质来控制,满足ICH Q3A、ICH Q3B要求即可。
三建立遗传毒性杂质检测方法
遗传毒性杂质限度较低,常用质谱检测器检测,如GC-MS、GC-MS-MS、HPLC-MS等灵敏度高的方法进行检测。但该类方法比较敏感,易受环境、仪器状态、样品基质状况、试剂纯度等影响,使用过程中需多加注意。
近年来,也有其他老师发表用衍生化法、荧光法等检测遗传毒性杂质的文章,大家建立方法时可以多查文献,集思广益。

来源:药研


