您当前的位置:检测资讯 > 生产品管
嘉峪检测网 2022-07-11 21:35
一般情况下,药品注册生产现场核查(以下简称生产现场核查)以技术转移所获取的知识为基础,以商业规模生产工艺验证批次为起点直至现场动态生产批次为止,重点包括商业规模生产工艺验证批次、动态生产批次以及在此期间的相关变更、稳定性试验等研究、试制的批次。
药品生产现场核查的目的主要是通过对申报品种的商业化生产条件和能力、数据可靠性进行实地核查,核实申报资料的真实性,核实商业化生产规模下相关生产和质量控制活动与申报资料(如处方、生产工艺、质量标准、关键设施设备等)的一致性以及商业化生产条件。
本文对药品注册生产现场核查注意事项及常见缺陷进行列举,供业内同仁参考。
一、检查工作流程
药监方面 企业方面
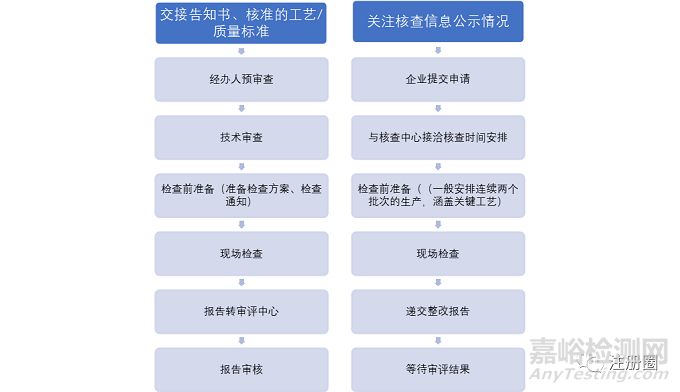
二、需提前准备的现场检查文件清单
1.组织结构图
2.质量部门和质量体系
3.企业许可证和GMP检查概况
4.生产车间平面图
5.相关操作间和关键设备分布图
6.操作人员、物料、中间产品、成品和废料处理的流向图
7.生产工艺流程图
8.生产车间其他产品情况
9.委托生产和委托检验清单及相关委托协议
10.关键生产设备清单,包括直接接触和不直接接触产品的设备
11.物料供应商清单
12.相应品种涉及的关键检验设备的清单
13.相应品种涉及的SOP清单,包括工艺规程和检验规程等
14.相应品种所有生产批/批次的批生产记录
15.变更控制、偏差、OOS清单
16.其他检查组要求的检查相关文件
三、技术审查常见问题
1.生产地址发生实质性变更;
2.原料药或关键辅料发生重大变更;
3.工艺变更;
4.生产工艺验证三批不是连续生产的三批;
5.验证三批批量不同,小于上市批量且不在生产线允许范围内,缺少科学评估基础。
四、现场检查常见缺陷
(一)质量管理
药品生产企业应当具备涵盖影响药品质量所有因素的质量体系,具有与药品生产相适应的组织机构,并建立质量保证系统以保证质量体系的有效运行。
常见缺陷示例:
1.人员培训不到位,如检验人员对方法设置、方法调用等操作功能不熟悉;未查见预混、总混工序的现场操作人员及现场QA对检查品种的生产工艺规程、批生产记录等的相关培训记录。
2.相应操作SOP无生效版本,相关人员未经过SOP培训。
3.偏差纠正预防措施不到位,如验证批12个月稳定性样品的聚合物检测出现偏差,调查原因显示为方法设置和调用错误,预防措施为建立方法后专人复核,但未制订具体的复核方法。
(二)厂房与设施、设备
企业的厂房、设施、关键生产设备应当与注册申报资料一致,并与商业化批量生产匹配,药品生产过程中防止污染与交叉污染的措施应当有效。
常见缺陷示例:
1.厂房、设施、关键生产设备的设计确认、安装确认、运行确认和性能确认不完整,无法满足样品商业化批量生产要求。
2.非专用生产线的共线评估不完整,清洁验证不完整,如未对活性物质残留量/积累量进行评估。
3.计算机系统未经过确认,不符合计算机化系统管理规范。
4.仪器仪表未在校验效期内。
5.洁净器具的管理不规范。
6.设备标识牌未区分“待使用状态”、“待清洗”、“已清洗状态”等不同状态。
(三)物料
涉及相关物料的采购、接收、贮存、检验、放行、发放、使用、退库、销毁全过程,应当确保物料在上述过程不发生污染、交叉污染、混淆和差错。原辅料和直接接触药品的包装材料和容器的质量标准、生产商/来源应当与注册申报资料一致,按照相关标准操作规程进行取样和检验,并出具全项检验报告。
常见缺陷:
1.现场未查见对产品生产所用的原辅料、直接接触药品的包装材料和容器的供应商的审计记录和管理规程。
2.生产所用的原辅料和直接接触药品的包装材料和容器的质量标准、生产商/来源与注册申报资料不一致。
3.原辅料发放操作与标准操作程序不符。
4.物料贮存条件与质量标准不一致。如薄荷粉末香精及香蕉粉末香精的质量标准中规定,“应贮存在阴凉、干燥通风的仓库内”,实际贮存于常温库中。
(四)批量生产
以商业规模生产工艺验证为起始,确认企业生产工艺与注册资料的一致性,以及持续稳定生产出符合注册要求产品的能力。
常见缺陷示例:
1.未按要求进行批量生产工艺验证且缺少科学、合理的解释。
2.工艺流程及各项工艺参数未确定。
3.商业规模生产工艺验证批及现场动态生产批次的处方、批量、实际生产过程、批生产记录与工艺规程/制造检定规程和注册申报工艺不一致,如1)工艺规程批量确定为最小批量10000瓶,而工艺验证三批的理论批量分别为0.88、0.85、0.81万支;2)工艺规程要求成品率为93%--105%,批生产记录中三批成品率分别为81%、82%、83%,未开展偏差调查,批生产记录中未备注原因;3)现场查见洗瓶工序中的纯化水压力、压缩空气压力和注射用水压力均超出工艺规程中的工艺控制点范围;4)商业规模生产工艺验证批与现场动态生产批次生产所用设备不一致;5)申报资料中生产工艺为羧甲淀粉钠过40目筛后直接投料,而工艺验证批与动态批生产中羧甲淀粉钠在投料前90℃干燥4小时再过40目筛;6)申报资料中,铝塑包装后的产品采用药品包装用复合膜袋进行热封,并放入干燥剂,工艺验证批、现行工艺规程和动态批生产中未见放入干燥剂;7)申报资料中,制粒工序为“将活性成分与辅料乳糖、微晶纤维素和羧甲淀粉钠采用等量递加法混合,40目过筛两遍使混合均匀,加入适量的粘合剂4%HPMC 95%乙醇溶液制得软材”,动态批生产中采用高效混合制粒机一步混合制粒。
4.动态生产批分装工序装量调节时,操作人员多次直接打开A级层流罩在分装机附近取样,未从指定的取样点取样。
5.生产工艺规程中成盐工序所用试剂注明“回收套用”,但未对回收套用上述溶剂进行验证。
6.工艺验证不充分,如工艺验证方案关键参数风险评估表中,压片工序的转速和压力为关键工艺参数,验证报告中仅对转速进行验证,未对影响产品的硬度、脆碎度的压力作验证。
7.动态生产批发生偏差,未启动偏差调查,未在批生产记录中记录偏差相关信息,制定的纠正预防措施不合理等。
(五)质量控制
质量控制实验室的人员、设施、设备应当与产品质量控制相适应,应当配备药典、标准图谱等必要的工具书,以及相应的标准品或对照品等相关标准物质。企业应当建立相应质量控制制度,按药品生产质量管理规范要求进行取样、检验,并得出真实可靠的检验结果。
常见缺陷:
1.检验设施设备仪器未经过检定或校准,或已过有效期。
2.样品、中间产品/中间体和关键物料的质量标准与申报的质量标准存在不一致。
3.检验方法未经过方法学验证,或验证内容不完整。
4.样品、标准物质、试剂、菌种等管理不规范,如对照品领用台账上,领用人与管理人均为同一人。
5.稳定性试验不规范,如实际留样量与台账登记不符,长期稳定性试验,记录为每批放置14盒,余10盒,实余量为每批11盒;加速稳定性试验,记录为每批放置8盒,余0盒,实余量为每批2盒。批生产记录中显示,每批取留样26盒,其中每批常温留样4盒,实际常温留样为每批2盒。
6.未对有存放效期的中间产品/中间体进行相应稳定性研究。
7.委托检验,未对受委托方进行审计,未签订合同或协议,或签订日期晚于检验日期。
8.计算公式有误,如成品检验操作规程含量测定项下,规定“取原料药钠盐对照品适量”配制对照品溶液,计算结果以原料药游离碱计,计算公式中未体现游离碱与钠盐的换算校正因子
9.微生物限度检查与现行《中国药典》四部通则的要求存在不一致,如1)药典中规定供试品检验量“一般应随机抽取不少于2个最小包装的供试品,混合,取规定量供试品进行检验”;检验人员实际取一个成品包装内的10ml进行检验。2)药典中规定菌数报告规则为“若滤膜上无菌落生长,以<1报告菌数”,微生物检验报告中未发现菌落生长的样品报告为“0”。3)清洁验证取样样品进行微生物限度检查前,未进行微生物计数方法适用性试验。
(六)数据可靠性
企业应当采取有效措施防止数据的修改、删除、覆盖等,以确保数据可靠。申报资料中的数据均应当真实、准确,能够溯源,相关的原始记录、原始图谱、原始数据等均应当与申报资料一致。其中,商业规模生产工艺验证及其稳定性试验等生产、检验数据尤为重要。
常见缺陷示例:
1.原始记录不完整,不清晰,如打印纸已褪色,批记录中填写不完整,缺少操作人员签名等。
2.计算机系统管理不规范,如:
1)高效液相色谱仪的计算机权限设置了操作员、分析员和实验室负责人三级权限,实验室负责人权限涵盖操作员和分析员权限且实际参与部分检测,但实际未对实验室负责人的电子数据进行复核,也未明确复核人的资质;
2)原子分光光度计存放数据的文件夹未设置禁止删除权限,仪器使用记录中未体现数据备份信息。

来源:注册圈


