您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-06-14 05:47
药物的固有溶出(apparent intrinsic dissolution)是指一定量的药物(一般指原料药)在一定介质中,单位面积和单位时间内药物溶出的质量。通常表示为固有溶出速率(intrinsic dissolution rate,IDR),其值以mg·cm-2·min-1表示。
药物的固有溶出不同于溶出度。固有溶出是药物理化性质的重要参数之一,是药物固有的特性;而溶出度是指药物在规定介质中从药物制剂溶出的速度和程度,受到制剂辅料、制剂类型、工艺、纯度、介质等因素的影响,不能反映药物自身的特性。
同时,固有溶出也不同于溶解度。
固有溶出反映的是药物溶解的速度和程度,与体内溶出动力学密切相关;而溶解度只能反映化合物的溶解程度。
固有溶出可以在一定程度上反映药物的纯度,也可以用于评价原料药不同来源、批次间、不同结晶条件或不同晶型的一致性,还可用于考察药物不同盐基、晶型、粒度、表面活性剂、pH值等因素对溶出的影响,在一定程度上预测药物制剂的溶出行为,从而预测药物的生物利用度和疗效。
因此,药物的固有溶出速率可以为原料药质量评价、生产工艺的筛选提供数据支撑,为药物剂型的选择和处方的设计提供指导意见。
药物固有溶出的测定方法主要有2种:圆盘法和颗粒法。
圆盘法是将一定量的药物压制成薄片,放入溶出介质中,测定单位面积和单位时间内药物溶出的质量。
根据圆盘放置位置又可以分为转盘法和定盘法。由于圆盘法测定结果比较稳定,使用范围比较广,所以被多国药典所收载。
颗粒法是将一定粒径的药物原料颗粒放置在溶出介质中,测定单位时间内药物溶出的质量。
根据溶出介质是否含有助悬稳定剂,颗粒法又分为粗颗粒法和悬浮粒子法。
本文将重点对转盘法、定盘法、粗颗粒法及悬浮颗粒法的试验方法、影响因素及应用情况进行介绍。4种固有溶出测定装置示意图如图1所示。
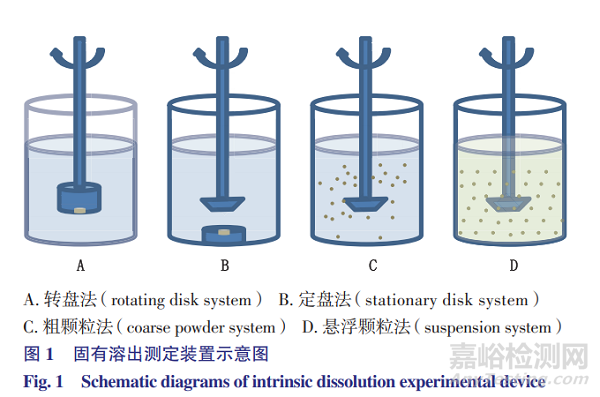
圆盘法
圆盘法是采用适宜的装置将药物压成直径固定的非崩解薄片,将药物薄片及载药模具一起放入一定量的溶出介质中,调整模具及搅拌桨的位置,在一定的转速下、特定的时间点取样分析。
圆盘法最早可以追溯到20世纪50~60年代John Wood开发的“Woods Apparatus”转盘法实验装置(rotating disk system)。随后Viegas和StelleAustin等人又发明了定圆盘实验装置(stationary disksystem)。
转盘法是载药模具旋转,溶出介质静止不动;定盘法是载药模具静止不动,通过搅拌桨或磁力搅拌器使溶出介质流动。在试验过程中药物薄片应不崩解、不剥离、不脱落,圆盘表面积固定,模具在容器中的位置固定,以降低流体动力学条件的影响。
目前,美国药典(USP42-NF37 1087)、欧洲药典(EP9.0 2.9. 29)和英国药典(BP2019 Appendix XII B5)均收载了圆盘固有溶出测定法。其中USP42-NF37中收载了转盘法和定盘法,并规定了测定方法、实验装置、实验参数及数据处理办法,包括药物压片参数、样品位置、搅拌桨位置、搅拌速度范围等。
EP9.0和BP2019内容基本一致,仅收载了转盘法,且未对转速提出建议和要求。到目前为止,我国包括《中华人民共和国药典》在内的药品标准均没有收载固有溶出测定法。
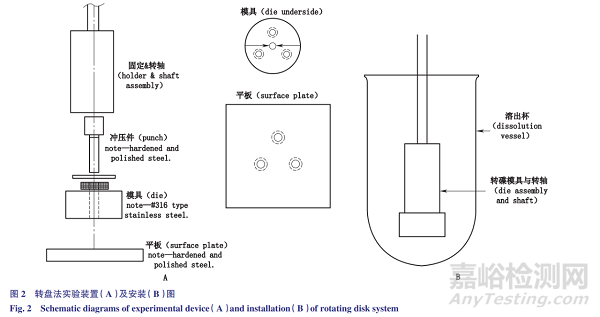
转盘法:转盘法实验装置见图2-A。药物薄片被压制在的旋转支架下部的模腔中,药物薄片下表面暴露。将载药模具浸入溶出介质中(图2-B),设定转动速度,进行试验。实验参数包括:药物用量<500mg;压力:15MPa,保持1~2min;转速:60~500r·min-1(优选300r·min-1);距底部的距离:不低于1.0cm;溶出介质体积:225~900mL;模具的直径:0.1~1cm。
定盘法:转盘法实验装置见图3-A。药物薄片被压制在定盘模腔中,药物薄片上表面暴露。将载药模具放置在平底溶出杯底部(图3-B)。搅拌装置(如桨叶)与药物薄片的距离通常为2.54cm。
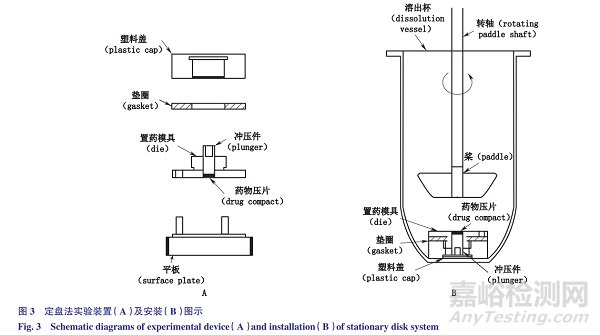
转盘法和定盘法相似之处在于:采用标准的溶出仪装置,药物薄片表面积恒定,在实验过程中药物薄片不碎裂和脱落,只有一个表面暴露在溶媒中,载药模具在溶出杯中的位置固定,旋转速度保持恒定,以减少水流动力变化的影响。
不同之处在于:液流动力来源不同,转盘法的液流动力来源为模具的转动,而定盘法液流动力来源是桨或其他搅拌设备的转动。将药物溶质的累积量与时间作图,即可得到固有溶出速率。
药物浓度随时间曲线的向上曲率(正二阶导数)通常提示实验系统存在问题,如因压片开裂、分层或溶解而导致了样品崩解;曲线的向下曲率(负二阶导数)通常表示薄片上药物的固体形态发生了变化,如热力学不稳定的晶型转变为更稳定的晶型,或者溶质在溶出介质中接近饱和。
在溶剂、温度、实验室设备和实验设置等试验参数一致的情况下,圆盘法的测定结果是一个相对稳定的数值。该数值对药物成药性研究具有非常重要的意义。
有研究表明,若某药物的固有溶出速率大于1mg·cm-2·min-1,说明该药物在制备成口服固体制剂时,其溶出过程一般不会成为吸收的限速过程;若小于1mg·cm-2·min-1,则在开发口服固体制剂时,需要采取一定的方法增加其溶解度或溶出速率,如成盐、包合物、固体分散体、共晶等。
圆盘分析法适用于表观溶解度>1mg·mL-1的化合物,对于难溶性药物的圆盘法测定耗时较长。
此外,在试验过程中圆盘法需要消耗的样品量比较大,一次测定通常需要约500mg的样品和900mL的溶出介质,即便是微量圆盘法也需要5mg左右的样品才能进行测定。
在新药研发初期,需要筛选的候选化合物种类较多,各候选药物的样品量非常少,但是还迫切需要参考固有溶出速率数值,颗粒分析法应运而生。
颗粒法
颗粒法是将一定粒径的药物原料颗粒放置在溶出介质中,测定单位时间内药物溶出的质量。
颗粒法需要的样品量大大减少(约10mg),通过减小溶出介质的体积,颗粒法样品量可以减小到几毫克或更少。同时由于颗粒与溶解介质接触表面积远大于圆盘法,因此可以将难溶性药物固有溶出速率测定时间从几小时缩短到几分钟,非常适合于药物研发初期大量候选化合物的筛选。
通常情况下,圆盘法适用于表观溶解度>1mg·mL-1的化合物,通常在60min内得到溶解曲线。而颗粒法法适用于表观溶解度<100μg·mL-1的化合物。两者之间的化合物可以用这2种方法中的任何一种来研究。
粗颗粒法:
粗颗粒法测定药物固有溶出,是将一定粒径的药物粗粉直接称重到试验容器中,并添加溶解介质(通常预热至37℃),测定单位时间内药物溶出的质量,根据颗粒比表面积、形状或尺寸分布计算固有溶出速率。
有研究表明,粗颗粒法的测量速度比圆盘分析法快约100倍,且与圆盘法获得的数据有良好的一致性。
对于任何给定的药物,通过减小颗粒粒径增加固体药物的总表面积,其溶解速率将成比例地增加。但是,随着粒径的减小,药物在溶出介质中易产生聚集效应,聚集效应会减小样品颗粒与溶解介质的接触面积,从而影响固有溶出测定结果。因此粗颗粒法应特别注意对粒径的控制,避免药物颗粒聚集。
此外,颗粒的孔隙率或药物在介质中的中分散性,也会影响到固有溶出测定结果。
也有文献表明,在使用粗颗粒法测定固有溶出速率的过程中,不需要测定颗粒粒径及表面积,也可以进行分析。
分析过程中经常会应用到NoyesWhitney药物溶出速度方程、Nernst-Brünner溶出动力学方程、双组分球形颗粒方程和WangFlanagan非沉降球形颗粒方程等多种数学公式,大量数学公式的使用反而限制了颗粒法在药物研究中的推广应用。
悬浮颗粒法:
悬浮颗粒法是将一定粒径的固体药物分散在一定量的分散介质中,形成高浓度悬浮液,再将该混悬液加入到一定介质中,测定单位时间内药物溶出的质量。
采用激光粒度仪或动态光散射测定仪测量悬浮液的粒径,颗粒总表面积可以通过计算得到。
分散介质中通常通过添加低浓度聚合物和/或表面活性剂形成分散良好、稳定的悬浮液(若发生沉淀,可振摇再分散形成悬浮液)。由于悬浮颗粒法不容易产生药物颗粒聚集,因此比粗颗粒法更有优势。
与圆盘法或粗颗粒法相比,悬浮颗粒法的主要优点是样品比表面积较大,溶解速度快,可快速测定。悬浮颗粒测定法要求分样品颗粒分散良好,无团聚物。
有研究表明,采用悬浮颗粒法测定了一系列粒径约为1μm的难溶化合物的固有溶出速率,其测定结果与圆盘法呈现显示良好的一致性。
需要注意的是,悬浮颗粒测定法要求样品粒径不能太小,太小的颗粒如纳米颗粒的悬浮液在测定过程中经常会出现团聚现象。
悬浮颗粒可以使用搅拌研磨机、球磨机、超声波发生器或高压均质机等方法制备。
悬浮介质通常采用低浓度的表面活性剂和/或聚合物做稳定剂,通过产生静电排斥或空间位阻稳定颗粒,防止颗粒团聚。
常用的稳定剂是采用羟丙基甲基纤维素和聚乙烯吡咯烷酮等聚合物与十二烷基硫酸钠等表面活性剂结合使用。
较高浓度的稳定剂可以提高悬浮液的稳定性,但是由于表面活性剂可能会改变悬浮颗粒表面扩散层的厚度和表面流体动力学,而聚合物对颗粒的吸附可能会减少悬浮颗粒与溶出介质接触的比表面积,从而降低溶解速率,影响固有溶出测量结果。
因此,为减少表面活性剂和聚合物对固有溶出测定结果的影响,试验中因尽可能选择较低浓度的稳定剂。
其他测定方法
由于传统的圆盘法测定药物的固有溶出速率需要样品量较大,约500mg,而药物早期研发筛选阶段的样本量仅有几个毫克。为了解决这一矛盾,在圆盘法、颗粒法的基础上,研究人员开发使用了小型圆盘法、微量悬浮颗粒法、原位光纤分析技术、流通池法、表层溶蚀成像法、紫外成像溶出度测定法、单粒子光学成像法等方法,用于微量样品固有溶出的测定。
例如,研究者在小型圆盘法和微量悬浮颗粒法中采用原位光纤技术,使用相对少量的原料药,即可得到与传统圆盘法相一致的固有溶出速率数据。
Svanbäck等人开发的单粒子光学成像方法,仅用几毫克的原料药就可以得到固有溶出速率数据。Ward等开发的表面溶蚀成像技术,使用约3~10mg的原料药即可测定固有溶出速率。
影响因素
影响药物固有溶出速率的因素较多,内在因素包括:晶型、结晶度、无定型态、润湿性、粒度、比表面积、共沉淀物以及杂质含量等;外在因素包括:流体动力学、溶解介质性质(包括溶剂、表面活性剂、温度、流体粘度、pH、缓冲液类型和强度)、实验参数(如实验装置类型、搅拌桨转速等,还有压片参数,如药物用量、压片直径、压力和压制时间等)。
溶出介质的选择
介质体积应满足漏槽条件,以避免因接近饱和而降低溶出速率。
固有溶出通常在水介质中进行,为了接近体内条件,也可在37℃生理pH范围内测量。水应在脱气后立即使用,避免在片或模具表面形成气泡。
当样品为可电离化合物和盐时,应严格控制介质温度和pH,因为可电离化合物和盐的溶解速度主要依赖于溶液pH、缓冲液种类和缓冲液浓度。
当样品为酸性和碱性物质时,溶质溶解过程中可改变压片表面及附近的pH,导致压片表面pH与溶出介质本身pH相差很大。
为了消除压片表面溶质缓冲、溶液pH变化的影响,对于弱酸性化合物,溶出介质的pH应低于化合物pKa的1~2个pH单位,对于弱碱性化合物,溶出介质的pH应高于溶解物pKa的1~2个pH单位。
压片参数的选择
大部分药物采用液压机15MPa压制1min,即可得到试验用非崩解片。
压制成的薄圆片表面应完全完整平滑,无坑无裂,不能有松散粘附的粉末,并且溶解必须以相同的速率发生在整个圆盘上。
在溶解实验过程中,粗糙的表面会改变暴露在溶解介质中的总表面积,导致无法准确计算固有溶出速率。
在压片过程中,药物的晶型可能发生变化,可通过粉末X射线衍射或其他技术确认固态形态。
在压制过程中应注意避免形成毛细管或裂片,压片以后应在表面上吹压缩空气或氮气,除去药片表面的松散粉末。
颗粒的团聚效应
在粗颗粒法和悬浮颗粒法中,要求样品粒径不能太小,太小的颗粒如纳米颗粒在溶出介质中经常会出现团聚现象,从而改变样品颗粒与溶解介质的接触面积,影响固有溶出测定结果。
应用与展望
固有溶出是药物理化性质的重要参数之一,是药物的固有特性。固有溶出速率与溶解平衡无关,而是与溶解速率有关,比溶解度更能反映药物的溶解性能。
相比较溶解度而言,固有溶出速率与体内溶出动力学的相关性更大。因此,药物的固有溶出速率可以代替溶解度,用于药物在生物药剂分类系统中溶解性的判断,为新药研发提供参考。
固有溶出是药物研究过程中需重点关注的参数。
固有溶出速率可以反映原料药的化学纯度、晶型纯度,可作为药物质量控制手段用于药物生产过程;可用于考察药物不同盐基、晶型、粒度、表面活性剂、pH等因素对溶出的影响,用于优选原料的晶型、盐基,及筛选药物辅料;也可以用于评价不同来源、批次、晶型的原料药的差异,用于药物的一致性评价;还可以在一定程度上预测药物制剂的溶出行为,预测潜在的生物利用度等。
在创新药物研发中,通过对候选药物固有溶出测定,预测成药性,指导药物剂型选择和处方设计,提高新药研发的成功率和效率,避免资源浪费。如药物的固有溶出速率大于1mg·cm-2·min-1,说明溶出不会成为吸收的限速过程,可制备成口服固体制剂;反之,则需要采取一定的增溶方法,增加其溶解度或溶出速率。
随着技术的不断发展和完善,药物固有溶出测定法的应用会继续朝着更加微量化、准确化、智能化的方向发展,固有溶出测定技术在药学研究中的应用前景也将更加广阔。

来源:《药物分析杂志》


