您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-01-21 23:40
除菌过滤是指采用物理截留的方法去除液体或气体中的微生物,以达到无菌药品相关质量要求的过程[1]。除菌过滤器则是指用挑战水平大于等于1×107cfu/cm2过滤面积浓度的缺陷短波单胞菌对过滤器进行挑战,经过适当验证,可以稳定重现产生无菌滤出液的过滤器[2]。
在无菌药品生产过程中,尤其是对于非终端灭菌产品,主要依靠除菌过滤和无菌生产工艺来达到无菌控制要求[3]。随着除菌过滤技术在制药行业的广泛应用,除菌过滤的风险也引起了行业的重点关注。为加强药品生产监管,规范药品生产企业科学系统地开展除菌过滤技术及应用,保证无菌药品的安全、有效和质量稳定,国家药品监督管理局组织制定了《除菌过滤技术及应用指南》,作为实施《药品生产质量管理规范(2010年修订)》的指导性文件,自2018年10月1日起施行。
为正确理解《除菌过滤技术及应用指南》的技术要求,了解企业对指南的贯彻执行情况,把握除菌过滤过程中存在的问题,帮助企业规范应用除菌过滤技术,本文统计分析了近三年山东省药品生产检查中除菌过滤方面存在的主要问题并提出了解决对策。
1、 检查基本情况
2017年1月1日至2019年12月31日山东省共完成无菌药品生产企业检查159家次,其中无菌制剂124家次,无菌原料药35家次。无菌药品生产企业中存在除菌过滤缺陷的企业为72家次,占比45%,其中无菌制剂生产企业的除菌过滤缺陷比例高于无菌原料药。这一方面说明无菌制剂的除菌过滤问题更突出,另一方面也说明检查员对无菌制剂的除菌过滤风险关注度更高。检查企业和除菌过滤缺陷统计情况见表1。
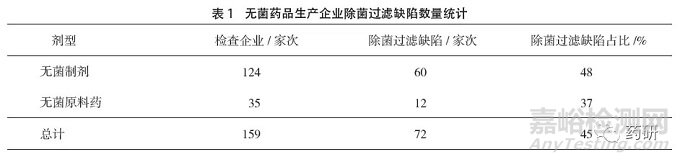
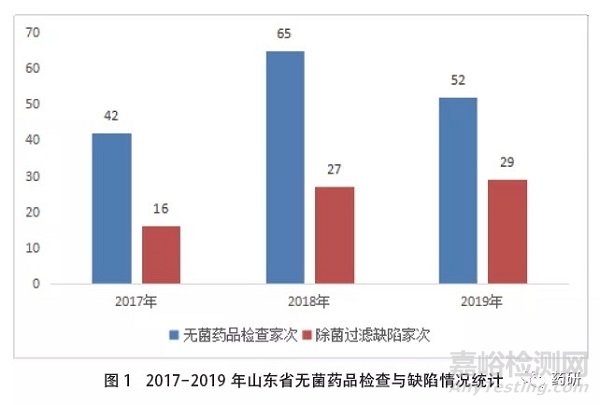
从各年度检查情况来看,2017-2019年度除菌过滤缺陷企业占无菌药品检查企业数量的比例依次为38%、42%、56%,呈现出明显的增加趋势,这可能与《除菌过滤技术及应用指南》施行后业内对除菌过滤的关注度提高和贯彻落实有关,同时也提示企业在无菌药品生产中要更加重视除菌过滤技术的规范应用。各年度缺陷情况统计见图1。
2、 除菌过滤问题统计
根据《除菌过滤技术及应用指南》的内容并结合无菌药品检查中除菌过滤缺陷项目,本文将除菌过滤技术及应用中的问题分为过滤工艺及系统设、除菌过滤验证、除菌过滤器和系统的使用、文件与记录4个部分,其中每个部分又分为若干个方面,对除菌过滤的缺陷内容分别依据缺陷部分和缺陷方面进行分类统计,统计情况见表2。

从表2中可以看出,各缺陷方面频次从高到低依次是使用、记录、完整性测试、过滤工艺的设计、重复使用、除菌过滤验证、规程、过滤系统的设计、灭菌。除菌过滤器的使用、完整性测试方面的缺陷往往伴随有记录方面的缺陷,虽然这3方面缺陷数量占总缺陷的60%,但大多是由于工作不完善导致的一般缺陷,将这3方面缺陷统一归纳为除菌过滤器使用和记录规范性问题。过滤工艺的设计、重复使用、除菌过滤验证3方面的缺陷各占总缺陷的12%、11%、8%,虽然缺陷数量占比不高,但其缺陷等级大多被划分为主要缺陷,有些甚至是严重缺陷,因此应该引起重视。
基于以上分析,目前除菌过滤技术在药品生产应用中的问题主要表现在除菌过滤器使用和记录规范性、过滤工艺的设计、重复使用、除菌过滤验证4个方面。
3 、解决对策
3.1 重视使用环节,规范操作和记录
除菌过滤器的使用、完整性测试和记录在除菌过滤技术应用中存在问题比例最高,这些问题大多是由于操作不规范或者记录不完整造成的,这就提示企业要重视使用环节,规范操作和记录。
除菌过滤系统的使用主要包括组装、密闭性确认、过滤参数控制、完整性测试等内容。企业应制定过滤器组装的操作规程,在组装过程中避免污染,并且要确保组装滤器的密封性,组装完成后根据情况对滤器进行预冲洗和密闭性确认。在生产过程中应对影响除菌过滤效果的关键参数,如过滤温度、时间、流速、压差等,进行控制和记录。完整性测试应按标准操作程序进行,测试过程中关注测试参数的设定、润湿的操作流程、测试的气体等内容[4-5]。
建议企业要建立完善的操作规程和记录文件,规程内容要涵盖滤器的组装、使用、完整性测试的各个方面,记录的内容要完整、可追溯。在生产操作中,要按照操作规程规范操作、及时记录。对在过滤系统使用中出现的任何异常和偏差都应该进行记录,必要时启动偏差调查。
3.2 关注过滤工艺设计,合理确定工艺参数
过滤工艺设计方面存在的问题主要是部分过滤工艺过程参数未确定以及供应商管理不完善。
企业在选择过滤器时,往往仅对过滤器的材质、规格型号等进行了考虑,过滤压差范围、温度范围、过滤流速、灭菌条件等工艺参数则参照滤器说明书执行。实际上,企业应根据待过滤介质属性及工艺要求确定过滤工艺参数,并对工艺参数范围进行确认。
部分企业仅建立有原辅料的合格供应商名单,将除菌过滤器作为工器具或耗材来管理,未严格按照供应商管理的要求进行质量控制。实际上,企业应建立包含除菌过滤器在内的所有生产用物料的合格供应商名单,除菌过滤器作为对产品质量有直接影响的主要物料,应进行必要的审计并签订质量协议。
3.3 充分进行评估验证,规范重复使用次数
液体除菌过滤器的设计和制造一般是按单次使用考虑的,但在实际生产中,往往存在重复使用的情形。企业应在充分了解产品和工艺风险的基础上,对过滤器的重复使用进行评估和验证,并明确规定生产中允许重复使用的最大次数和最长时间[6]。
除菌滤器重复使用的评估和验证是目前企业工作中的薄弱点和难点,建议企业参照《除菌过滤技术及应用指南》中重复使用的风险因素,结合产品和工艺特点,对重复使用中多次清洗、多次灭菌、使用时间延长等最差条件造成的完整性缺陷、细菌穿透、可提取物增加、产品残留、滤器堵塞、组件老化等风险进行充分的评估和验证,根据验证结果规定重复使用次数,并在使用过程中持续监测。
3.4 评估过滤工艺风险,开展除菌过滤验证
除菌过滤验证包含除菌过滤器本身的性能确认和过滤工艺验证两部分,除菌过滤器本身的性能确认一般由过滤器生产商完成,过滤工艺验证则是针对具体的待过滤介质并结合特定的工艺条件来实施。过滤工艺验证一般包括细菌截留试验、化学兼容性试验、可提取物或浸出物试验、安全性评估和吸附评估等内容。如果过滤后,以产品作为润湿介质进行完整性测试,还应进行相关的产品完整性测试验证[7-8]。
目前,大部分企业仍是委托滤器生产厂家或检验机构进行过滤工艺验证,但委托验证往往不能涵盖所有的验证项目,如委托验证未进行浸出物试验、吸附试验等。建议企业在充分把握过滤工艺验证要求的基础上,评估过滤工艺风险,确定需要验证的项目和程度,在委托试验的协议中就具体的验证项目、验证方法、分析方法等进行明确,确保过滤工艺验证的完整性。在委托试验机构出具验证报告后,企业应对报告的适用性和符合性进行确认,并将验证报告纳入企业的文件体系。此外,由于吸附试验需要对过滤介质进行分析,委托试验机构可能不具备检测条件,那么企业应自行开展此项验证。
4 、讨论
除菌过滤技术对于无菌药品特别是非终端灭菌药品的无菌保障具有非常重要的作用。除菌过滤系统的设计、除菌过滤器的选择和验证、使用过程中的参数控制等因素都直接影响着药品的安全和质量[9]。因此,除菌过滤系统不仅是企业无菌药品生产过程中的关键控制点,同时也是药品生产检查的关注重点。
除菌过滤器使用和记录规范性、过滤工艺的设计、重复使用、除菌过滤验证是除菌过滤技术应用中问题较为集中的方面。过滤介质、工艺条件和除菌过滤器是影响除菌过滤效果的三个重要因素。无菌药品生产企业一定要根据产品和工艺特点,结合除菌过滤器的特性,合理确定过滤工艺参数,开展过滤工艺验证,规范使用操作并进行记录,使其达到最佳效果,最终确保无菌药品的生产质量。
正确应用除菌过滤技术能够使滤过液达到无菌的要求,但除菌过滤并不能作为保证产品无菌的唯一手段,应在除菌过滤后端的生产过程中采取无菌生产工艺控制微生物污染才能确保产品的最终无菌[10]。

来源:中国药事


