您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2020-06-05 17:32
今日,国家药监局发布肌腱韧带固定系统等5项注册技术审查指导原则的通告(2020年第36号),《全膝关节假体系统产品注册技术审查指导原则》正式发布,全文如下。
全膝关节假体系统产品注册技术审查指导原则
本指导原则旨在为注册申请人进行全膝关节假体系统产品的注册申报提供技术指导,同时也为医疗器械监督管理部门对注册申报资料的审评提供技术参考。
本指导原则系对全膝关节假体系统产品注册申报资料的一般要求,注册申请人应依据具体产品的特性确定其中的具体内容是否适用,并对注册申报资料的内容进行充实和细化。
本指导原则是对注册申请人和审查人员的指导性文件,但不包括注册审批所涉及的行政事项,亦不作为法规强制执行,如果有能够满足相关法规要求的其他方法,也可以采用,但是需要提供详细的研究资料和验证资料。应在遵循相关法规和标准的前提下使用本指导原则。
本指导原则是在现行法规和标准体系以及当前认知水平基础上制定的,随着法规和标准的不断完善,以及科学技术的不断发展,本指导原则相关内容也将进行适时的调整。
一、适用范围
本指导原则涉及的产品适用于全膝关节假体置换术,包括股骨部件(股骨髁、股骨延长柄)、胫骨部件(胫骨衬垫、胫骨托、胫骨延长柄、紧固件)、髌骨部件(一体式髌骨、髌骨托/髌骨衬垫)、膝关节填充块、袖套等,由YY 0502《膝关节假体》标准(注:本指导原则中所列标准适用最新版本,下同)中已认可的金属材料、高分子材料、陶瓷材料和涂层材料制成。本指导原则不包含对部分膝关节置换用假体、特殊设计或个性化定制的全膝关节假体的要求,但适用部分可参照本指导原则。
二、技术审查要点
(一)产品名称的要求
根据《医疗器械分类规则》(国家食品药品监督管理总局令第15号)、《医疗器械通用名称命名规则》(国家食品药品监督管理总局令第19号)等相关文件,按照申报产品的设计特征和适用范围,确定其管理类别、分类编码及规范性命名,并论述其确定依据。
(二)产品的结构和组成
全膝关节假体系统通常包括股骨部件(股骨髁、股骨延长柄)、胫骨部件(胫骨衬垫、胫骨托、胫骨延长柄、紧固件)、髌骨部件(一体式髌骨、髌骨托/髌骨衬垫)、膝关节填充块、袖套等。
(三)注册单元划分的原则和实例
为全面评价全膝关节假体产品的安全有效性,鼓励按照系统划分注册单元。
1.若按照系统进行申报,应根据产品的固定方式、固定原理、适应证,将产品划分为如下注册单元:
(1)骨水泥型全膝关节假体系统
(2)非骨水泥型全膝关节假体系统
(3)混合型全膝关节假体系统
2.若以同一系统内单个或多个组件(如膝关节填充块、袖套、延长柄等)为注册单元进行申报,须明确与该产品配合的组件名称、产品的固定方式等。
3.材质(如有涂层时,涂层差异亦应考虑)不同的同类组件应划分为不同的注册单元,作为单一整体组配或组合使用的组件可以按同一注册单元申报。如:不同材质的胫骨托应作为不同的注册单元申报;作为同一系统组件配合使用的、材质已确定且唯一的组件,如股骨髁(钴铬钼合金)、胫骨衬垫(超高分子量聚乙烯)、胫骨托(钛合金),虽组件间材质不同,但因组合使用,可作为同一注册单元申报。
4.主要制造工艺方法不同(如锻造、铸造、不同热源及工艺的增材制造;烧结、等离子喷涂等涂层制备工艺)的同类组件,应视为不同注册单元。
(四)产品型号规格及基本信息
1.产品组成以及每个组件的名称和型号;提供产品各型号规格的划分原则。产品各组件的材料牌号/成分及其符合的国家标准、行业标准、国际标准,材料牌号的描述应与符合的标准一致。
2.产品的表面改性处理情况及相关制备工艺,如多孔涂层、耐磨涂层、骨水泥结合界面的喷砂粗化处理等。
3.提供体现产品及各组件几何结构特征及技术特点的三维立体结构图,概述主要设计特点、假体的限制程度、适用病例的侧副韧带功能及是否保留前后交叉韧带等。
产品描述举例:
(1)前后交叉韧带保留型全膝关节假体(BCR, Bi-cruciate retaining total knee joint prosthesis)
(2)后交叉韧带保留型全膝关节假体(CR, Cruciate retaining total knee joint prosthesis)
(3)后交叉韧带替代型全膝关节假体(如:PS, Posterior stabilized total knee joint prosthesis;AS, Anterior stabilized total knee joint prosthesis;CS, Cruciate substituting total knee joint prosthesis; DD,Deep Dish knee joint prosthesis;)
(4)前后交叉韧带替代型全膝关节假体(BCS, Bi-cruciate substituting total knee joint prosthesis)
(5)髁限制型膝关节假体(CCK, Constrained condylar knee joint prosthesis)
(6)旋转铰链膝关节假体(Constrained Rotating Hinged Knee Prosthesis)
(7)纯铰链型膝关节假体(Hinged Knee Prosthesis)
4.产品典型结构示意图中明确重要尺寸的起止点以及可用尺寸范围的整体说明;明确产品的几何尺寸及表面粗糙度,如股骨深度和宽度、胫骨深度和宽度、胫骨衬垫厚度,髌骨部件厚度等识别产品规格特征、结构和配合的重要尺寸,建议参考YY/T 0924.1《外科植入物部分和全膝关节假体第1部分:分类和尺寸标注》标准的要求。
5.提供产品的适用范围、禁忌证,必要时提供相应的文献资料以论证其范围的合理性;膝关节假体之间的部件兼容性,不同型号规格之间的匹配使用情况说明。以部件进行申报的产品应明确与产品配合使用的部件,并对其匹配的合理性予以说明。
6.阐述申报产品的研发背景和依据,以及与参考产品(同类产品或前代产品)的比较。如参考的同类产品(国内外已上市)或前代产品(如有),应当提供详细对比信息,同类产品应当说明选择其作为研发参考的原因,列表比较说明产品与参考产品(同类产品或前代产品)在工作原理、结构组成、制造材料、性能指标、关节面几何尺寸特征、适用范围等方面的异同。
(五)产品适用的相关标准
相关国家标准、行业标准、国际标准等举例,包含但不限于如下(本指导原则中所列标准适用最新版本):
YY 0502《膝关节假体》
GB/T 10610《产品几何技术规范(GPS)表面结构轮廓法评定表面结构的规则和方法》
GB/T 14233.1《医用输液、输血、注射器具检验方法第1部分:化学分析方法》
GB/T 14233.2《医用输液、输血、注射器具检验方法第2部分:生物试验方法》
GB/T 16886《医疗器械生物学评价系列标准》
GB 18278.1《医疗保健产品灭菌湿热第1部分:医疗器械灭菌过程的开发、确认和常规控制要求》
GB 18279.1《医疗保健产品灭菌环氧乙烷第1部分:医疗器械灭菌过程的开发、确认和常规控制的要求》
GB/T 18279.2《医疗保健产品的灭菌环氧乙烷第2部分:GB 18279.1应用指南》
GB 18280.1《医疗保健产品灭菌辐射第1部分:医疗器械灭菌过程的开发、确认和常规控制要求》
GB 18280.2《医疗保健产品灭菌辐射第2部分:建立灭菌剂量》
GB/T 18280.3《医疗保健产品灭菌辐射第3部分:剂量测量指南》
GB/T 19701.1《超高分子量聚乙烯第1部分:粉料》
GB/T 19701.2《超高分子量聚乙烯第2部分:模塑料》
ASTM F2695《外科植入物用与α生育酚(维他命E)混合的超高分子量聚乙烯粉末的规范》
GB 23101.2《外科植入物羟基磷灰石第2部分:羟基磷灰石涂层》(ISO 13779-2)
YY 0117.1《外科植入物骨关节假体锻、铸件Ti6Al4V钛合金锻件》
YY 0117.2《外科植入物骨关节假体锻、铸件ZTi6Al4V钛合金铸件》
YY 0117.3《外科植入物骨关节假体锻、铸件钴铬钼合金铸件》
YY/T 0343《外科金属植入物液体渗透检验》
YY/T 0772.3《外科植入物超高分子量聚乙烯第3部分:加速老化方法》
YY/T 0772.4《外科植入物超高分子量聚乙烯第4部分:氧化指数测试方法》
YY/T 0772.5《外科植入物超高分子量聚乙烯第5部分:形态评价方法》
YY/T 0811《外科植入物用大剂量辐射交联超高分子量聚乙烯制品标准要求》
GB/T 3505《产品几何技术规范(GPS)表面结构:轮廓法术语、定义及表面结构参数》(ISO 4287)
YY/T 0924.1《外科植入物部分和全膝关节假体部件 第1部分:分类、定义和尺寸标注》(ISO 7207-1)
YY/T 0924.2《外科植入物部分和全膝关节假体部件 第2部分:金属、陶瓷及塑料关节面》(ISO 7207-2)
YY/T 0810.1《外科植入物全膝关节假体第1部分:胫骨托疲劳性能的测定》(ISO 14879-1)
YY/T0652《外科植入物材料的磨损聚合物和金属材料磨屑分离、表征和定量分析方法》 (ISO17853)
YY/T 1426.1《外科植入物全膝关节假体的磨损第1部分:载荷控制的磨损试验机的载荷和位移参数及相关的试验环境条件》(ISO 14243-1)
YY/T 1426.2《外科植入物全膝关节假体的磨损第2部分:测量方法》(ISO 14243-2)
YY/T 1426.3《外科植入物全膝关节假体的磨损第3部分:位移控制的磨损试验机的载荷和位移参数及相关的试验环境条件》(ISO 14243-3)
YY/T 1427《外科植入物可植入材料及医疗器械静态和动态腐蚀试验的测试溶液和条件》
YY/T 1552《外科植入物评价金属植入材料和医疗器械长期腐蚀行为的开路电位测量方法》
YY/T 0640《无源外科植入物通用要求》(ISO 14630)
GB/T 12417.2《无源外科植入物关节置换植入物特殊要求》
YY/T 0919《无源外科植入物关节置换植入物膝关节置换植入物的专用要求》(ISO 21536)
ASTM F1609《磷酸钙涂层性能要求》
YY/T 0988.2《磷酸钙涂层和金属涂层的剪切试验方法》
YY/T 0988.11《磷酸钙涂层和金属涂层的拉伸试验方法》
YY/T 0988.13《磷酸钙涂层、金属涂层和磷酸钙/金属复合涂层的剪切疲劳试验与弯曲疲劳试验方法》
YY/T 0988.1《外科植入物涂层用钴28铬6钼合金粉》
YY/T 0988.2《外科植入物涂层用钛及钛6铝4钒合金粉》
YY/T 0988.14《外科植入物用多孔涂层立体学评价的试验方法》
YY/T 0988.15《使用Taber研磨机测试金属热喷涂涂层耐磨性的试验方法》
ASTM F1223《全膝关节假体约束度评价的标准测试方法》
ASTM F1672《表面髌骨假体标准规范》
ASTM F1800《全膝关节假体金属胫骨托疲劳试验标准》
ASTM F1875《模块化植入物界面微动腐蚀试验的标准操作规程:股骨头内锥与柄椎界面》
ASTM F1814《组配式髋关节和膝关节部件评价标准指南》
ASTM F2083《膝关节假体标准规范》
ASTM F2722《活动平台膝关节假体的胫骨平台旋转限制评价标准方法》
ASTM F2723《活动平台膝关节假体的胫骨平台/胫骨衬垫抗动态分离的评价标准测试方法》
ASTM F2724《活动平台膝关节假体脱位评价的标准测试方法》
ASTM F2777《评价高屈曲条件下膝关节胫骨衬垫耐久性和变形试验方法》
ASTM F3161《闭合加载条件下全膝关节假体金属股骨部件有限元分析标准试验方法》
ASTM F3334《全膝关节假体金属胫骨部件有限元分析》
VDI 3824 Part 1-part 4《物理气相沉积(PVD)和化学气相沉积(CVD)硬质涂层》
VDI 3198《冷锻工具的涂层》(CVD, PVD)
ISO19227《外科植入物的清洁度--通用要求》
(六)产品的适用范围/预期用途
产品适用于退行性骨关节炎、创伤性关节炎或缺血性坏死、炎性关节病等各种终末期膝关节疾病,以及畸形的矫正、肿瘤切除、假体失败后重建、应用其它技术无法处理的骨折等情形,须行全膝关节假体置换时使用。
(七)产品的主要风险
根据YY/T 0316《医疗器械风险管理对医疗器械的应用》,充分识别膝关节假体的设计、原材料采购、生产加工过程、产品包装、灭菌、运输、贮存、使用等产品生命周期内各个环节的安全特征,从生物学危险(源)、对手术操作的危险(源)、组件或注册单元功能失效危险(源)、环境危险(源)、有关使用的危险(源)、由于功能失效、老化及存储不当引起的可能危险(源)等方面,对产品进行全面的风险分析,并详述所采取的风险控制措施。
提供产品决定上市前对其风险管理活动进行全面评审所形成的风险管理报告,并在报告中说明和承诺:风险管理计划已被适当地实施;综合剩余风险是可接受的;已有恰当的方法获得与本产品相关的包括出厂后流通与临床应用的信息。
风险管理报告应包括风险分析、风险评价、风险控制等产品风险管理的相关资料。至少应包括:
1.产品安全特征清单;
2.产品可预见的危险(源)及危险(源)分析清单[说明危险(源)、可预见事件序列(即危险成因分析)、危险情况和可能发生的伤害之间的关系];
3.风险评价、风险控制措施以及剩余风险评价汇总表。
风险管理报告及相关资料的要求可参考附录1。
(八)产品的研究要求
1.材料及产品性能研究
对于申报产品,注册申请人应对产品材料、部件表面状态、产品设计及相关静/动态性能、体外磨损等方面的性能进行研究,并提供其可接受的依据,必要时,须与已上市同类别产品相关性能进行对比,以证明其安全有效性。
(1)针对全膝关节假体产品使用的超高分子量聚乙烯材料,在满足YY0502、YY/T0811、GB/T19701等标准的前提下,生产企业或原材料供应商还需针对产品所用原材料选取表1中适用项目提供研究资料。
表1超高分子量聚乙烯材料相关性能
|
性能 |
普通超高分子量聚乙烯 |
高交联超高分子量聚乙烯 |
含抗氧化剂高交联超高分子量聚乙烯 |
推荐测试标准 |
|
|---|---|---|---|---|---|
|
密度 |
X |
X |
X |
ISO 1183-1 ASTM D792 |
|
|
拉伸性能 |
抗拉强度 屈服强度 断裂伸长率 |
X |
X |
X |
ISO 527-1 ASTM D638 |
|
抗冲击性能 |
简支梁冲击强度 |
二者选其一(仲裁时选取简支梁试验) |
|
|
ISO 11542-2 |
|
悬臂梁冲击强度 |
X |
X |
ASTM F648, Annex A1 |
||
|
形态学测试 |
X |
X |
X |
YY 0502 YY/T 0772.5 ISO 5834-5 ASTM F648, Annex A2 |
|
|
氧化指数 |
SOI OI |
X |
X |
X |
YY 0502 YY/T 0772.4 ISO 5834-4 ASTM F2102 |
|
反式亚乙烯指数(TVI) |
|
X |
X |
YY/T 0814 ASTM F2381 |
|
|
热性能 |
结晶度 熔点 |
|
X |
X |
YY /T 0815 ASTM F2625 |
|
极限载荷 |
|
X |
X |
YY/T 1430 ASTM F2183 ASTM F2977 |
|
|
疲劳裂纹扩展 |
|
X |
X |
ASTM E647 |
|
|
压缩模量 |
|
X |
X |
ASTM D695 |
|
|
溶胀度 |
|
X |
X |
YY/T 0813 ASTM D2765 or F2214 |
|
|
剩余自由基 |
|
X |
X |
电子顺磁共振波谱法(EPR);电子自旋共振谱法(ESR) |
|
|
加速老化后性能 |
拉伸性能 |
X |
X |
X |
YY 0502 YY/T 0772.3 ISO 5834-3 ASTM F2003 |
|
抗冲击性能 |
X |
X |
X |
||
|
氧化指数 |
X |
X |
X |
||
|
形态学测试 |
X |
|
|
||
对于高交联超高分子量聚乙烯材料,生产企业或原材料供应商需提供超高分子量聚乙烯类型、辐照类型(γ射线或电子束)、辐照剂量、交联后热处理工艺过程(如重熔或退火,时间和温度)、机械热处理过程的压缩比等资料。交联工艺对材料磨损性能的影响(如磨损率、磨损表面形貌、磨损失效模式、磨屑分析等)、生物相容性评价等。
对于含抗氧化剂高交联超高分子量聚乙烯,生产企业或原材料供应商需提供超高分子量聚乙烯类型、抗氧化剂的成分、抗氧化剂的添加方法(与粉末混合或型材渗透、工艺过程中添加的稳定剂和助剂以及残留)、辐照类型(γ射线或电子束)、辐照剂量、交联后热处理工艺过程(时间、温度,如重熔或退火)、机械热处理过程的压缩比等资料。此外,还需提交以下研究资料:抗氧化剂的浓度、抗氧化剂在材料中的稳定性(在加载和/或体液环境下,随着时间推移抗氧化剂可能逐渐析出)、抗氧化剂对材料磨损性能的影响(如磨损率、磨损表面形貌、磨损失效模式、磨屑分析等)、抗氧化剂对材料固化的影响(材料成型后的形态学分析)、生物相容性评价等。
(2)部件表面处理及性能研究
a.喷砂处理:如生产工艺中对部件采用喷砂处理,需提供喷砂工艺验证资料,宜考虑表面形貌、粗糙度、喷砂颗粒物残留等相关因素;对于采用骨水泥固定的假体,分析论证产品表面形貌对股骨髁和胫骨托与骨水泥固定强度的影响。
b.多孔涂层:对于非骨水泥型假体,需提供多孔涂层制备工艺流程图及关键工序的质量控制点,提供多孔涂层的成分、厚度、形貌分析、涂层与基体的结合强度、涂层制备工艺对基体材料的影响、骨长入情况等研究资料。
羟基磷灰石涂层应符合GB 23101.2的规定。
对于等离子喷涂金属涂层,应按照YY/T 0988.14标准规定涂层厚度、孔隙率和平均孔隙截距的要求,提供相应研究资料。并按照YY/T 0988.2、YY/T 0988.11、YY/T 0988.13 标准分别进行剪切试验、拉伸试验、剪切疲劳试验,并考虑对涂层的弯曲疲劳性能进行评价。按照YY 0502标准要求,剪切强度应不低于20MPa,拉伸强度应不低于22MPa,疲劳试验应达到107正应力循环不失效,并提供载荷的确定依据。按照YY/T 0988.15进行耐磨性能试验,磨损100个周期后,涂层质量损耗总值应小于65mg(按重量计)。
c.股骨髁部件关节面耐磨涂层及表面改性层:在金属股骨髁部件关节面上通过化学气相沉积(CVD)、物理气相沉积(PVD)、表面化学热处理等方法制备耐磨涂层或表面改性层,需提供涂层及表面改性层的制备工艺流程图及关键工序的质量控制点(如基体表面光洁度、涂层制备或表面改性工艺流程图和风险控制方法、清洗工艺验证、质控标准等)。耐磨涂层应提供涂层成分、物相组成、涂层结构(如梯度设计、层数、各层的成分、物相及厚度)、厚度、表面形貌、硬度、涂层与基体的附着力(如划痕法、洛氏压痕法),耐腐蚀性(如适用)、耐磨损性能(如磨损率、磨损表面形貌、涂层表面分析、磨屑分析)等研究资料;涂层测试方法可参照但不限于VDI 3824、VDI 3198等标准进行试验;疲劳及磨损性能研究时,还应考虑模拟环境介质、接触应力、磨损周次等,分析涂层磨损量、涂层结合强度、磨屑尺寸及生物安全性,论证涂层在体内长期植入的稳定性和安全性。表面改性层须针对以上适用部分提供相关研究资料。
(3)部件动态疲劳试验
对于产品相关部件的动态疲劳试验,注册申请人应依据其在各项试验中的工况分别选取最差情况开展试验;最差情况的选择应结合产品的预期性能、设计特点、相应测试方法,采用有限元分析或其他已验证的方法进行。部件动态疲劳试验包括:
a.胫骨托的疲劳性能
胫骨托的疲劳性能应按YY/T 0810.1的试验方法进行试验,5个样件中的每一个样件应在最大载荷900N 和107次循环次数下试验,且不发生失效,以确定其在相应的加载条件和循环载荷下的疲劳性能。采用有限元方法选取最差情况时,可参照ASTM F3334标准的方法进行分析。
b.股骨髁的疲劳性能
股骨髁部件的疲劳性能可采用适合的方法进行试验,以确定在相应的加载条件和循环载荷下的疲劳性能。推荐在胫骨-股骨屈曲角90°时,选取5个同规格样品在确定的疲劳极限载荷下经历107次循环次数均不发生失效,并提供疲劳极限载荷的可接受依据。必要时,应在不同的载荷水平下建立F-N曲线,并确定107次循环次数的疲劳极限。同一注册单元内最差情况的选择,可参照ASTM F3161标准中有限元方法进行分析选取。
c.胫骨衬垫的立柱疲劳性能
对于采用凸轮-立柱机制实现后交叉韧带替代型膝关节假体,注册申请人应结合产品设计和已公开数据,对立柱断裂的风险进行评估。必要时,提供胫骨衬垫完整的立柱剪切疲劳试验报告。推荐在不同的载荷下进行试验并建立F-N曲线,且在确定的疲劳极限载荷下至少1个样件完成107次循环而不发生失效,并提供疲劳极限载荷的可接受依据。请考虑不同屈曲角度下凸轮-立柱接触位置、凸轮-立柱结构设计和尺寸(不同规格)等影响因素,选择最差情况进行试验。
d.高屈曲下胫骨衬垫耐久性和抗形变能力
注册申请人如宣称产品具有高屈曲的特点,应参照ASTM F2777方法评价胫骨衬垫在高屈曲角度下的耐久性和抗形变能力。
(4)组合式部件连接可靠性
对于通过机械锁定结构连接的全膝关节假体部件(如胫骨托/胫骨衬垫、髌骨托/髌骨衬垫等),连接机构应具有足够的完整性以覆盖预期使用的负载范围(或合理的部分负载)。建议注册申请人参照ASTM F1814对组合式部件的连接完整性进行评估,并对结果的可接受性进行论证。必要时可通过静态和动态剪切试验、弯曲试验、拉伸试验以及其他可能的组合试验对连接机构的性能进行评价。
对于固定平台胫骨部件,注册申请人需通过静态剪切试验(前后方向和内外方向)、静态拔出试验等评价胫骨托与胫骨衬垫之间的锁定结构强度。
对于活动平台胫骨部件,带有旋转限制结构胫骨假体应参照ASTM F2722标准对旋转限制结构的稳固性进行评估,选取5个样件并在最大扭矩14Nm和220000次循环次数下试验。可参照ASTM F2723标准对活动平台胫骨托/胫骨衬垫进行动态脱离试验。
对于组合式髌骨部件,注册申请人宜通过静态拔出试验和动态剪切试验评价髌骨托与髌骨衬垫之间的连接结构强度。动态剪切试验推荐在不同的载荷下选取样件进行,并建立F-N曲线,且至少一个样件完成107次循环而不发生失效。
微动腐蚀:对于组合式的部件,注册申请人可参照ASTM F1875标准对组件之间连接、微动和腐蚀进行评估,并对结果的可接受性进行论证。
(5)与设计相关的试验
a.相对角运动范围
注册申请人应按照ISO 21536标准规定的股骨部件和胫骨部件之间的相对角运动范围进行评估。相对角运动范围的评估应包含所有旋转模式(屈曲/伸展、内旋/外旋、内翻/外翻)和位移模式(向内/向外、向近端/向远端、向前/向后)下的数据。股骨部件和胫骨部件之间的相对角运动范围可通过植入物的CAD图纸进行估算。注册申请人需阐述如何定义中立位(包括股骨部件、胫骨部件在六个自由度的相对位置关系),0°屈曲角及屈曲伸展、内旋外旋和内外倾运动旋转轴位置。
b.约束度研究资料
根据ASTM F1223测定膝关节内外旋转、前后位移和内侧-外侧位移的全膝关节置换假体的约束能力。建议在0°、15°、90°和最大屈曲时对植入物进行分析、测试。ASTM F1223包含了对活动平台膝关节假体的特殊规定,并考虑关节下表面和整个植入物的上下关节面的约束能力测定。
系统在最大屈曲角时,应能在预期的生理载荷条件下允许± 15°内旋-外旋且不发生半脱位。部分特殊设计的限制型膝关节假体可能不满足上述要求,但应论述其合理性。
对于活动平台假体,可参照ASTM F2724的试验方法,对抗脱位能力(旋出或滑出)进行评估。
(6)全膝关节磨损试验
全膝关节假体的磨损试验应按ISO 14243-1或ISO 14243-3、ISO 14243-2标准要求进行,提供完整的实验报告,包括产品说明、试验设备描述、载荷和运动输入输出曲线、浸泡实验结果、液体介质(抗菌剂选用、润滑液体积、更换周期、变质情况说明等)、各个样品的磨损量和磨损率、关节面磨损区域和形貌等。对于实验过程中与ISO标准不一致的操作、参数设定等给出详细说明及依据,应与产品设计理念、假体运动学特征、宣称相对应。磨损试验中最差情况产品的选择,宜考虑假体类型、设计特点、测试样品在申报产品族中的代表性、试验加载方式(力控制和位移控制)、规格尺寸、关节面的接触应力、接触面积、滑动距离等影响因素,提供确定的依据。
对于新型的关节面材料、耐磨涂层或关节曲面设计,均应进行磨损试验评估,对磨损量、磨损率、关节面磨损区域和形貌、磨屑的形态和尺寸分布进行表征,可参考YY/T 0652标准,必要时须进行生物相容性评价;并将其与已成功应用的全膝关节假体数据进行比较分析,评价其临床安全性和有效性。
(7)MRI兼容性测试
注册申请人如对申报产品进行MRI兼容性的相关验证,可根据YY/T 0987系列标准对产品在核磁环境下的磁致位移力、磁致扭矩、射频致热、伪影等项目进行评估。并根据研究报告,列出MRI试验设备、磁场强度、比吸收率(SAR)等试验参数及温升、位移力、扭矩及伪影评估结果,相关信息在说明书中予以明示。
注册申请人如未对申报产品进行MRI兼容性的相关验证,应重点明确该产品尚未在MRI环境下对该产品的温升、移位状况及伪影进行测试评估。并在说明书的警示中注明相关内容,提示其存在的风险。
2.对于膝关节填充块、袖套等部件,如采用增材制造工艺成型,还需满足《无源植入性骨、关节及口腔硬组织个性化增材制造医疗器械注册技术审查指导原则》(国家药品监督管理局通告2019年第70号)中的相关要求。
3.生物相容性评价
产品的生物相容性评价应涵盖终产品及其涂层充分考虑磨损、微动腐蚀等因素的影响,并参照GB/T 16886.1-2011《医疗器械生物学评价第1部分:风险管理过程中的评价与试验》中的系统方法框图及《国家食品药品监督管理局关于印发医疗器械生物学评价和审查指南的通知》(国食药监械〔2007〕345号)中的审查要点进行风险评价,在缺乏相关数据时,补充进行必要的生物相容性测试。
4.产品灭菌确认
对于经辐照灭菌的产品,需明确辐照剂量及相关的验证报告,具体的剂量确定依据可参照GB 18280系列标准。
对于经环氧乙烷灭菌的产品,需提供灭菌结果确认和过程控制报告,具体可参照GB 18279系列标准。
5.产品有效期(货架寿命)的验证资料
灭菌产品应参照《无源植入性医疗器械货架有效期注册申报资料指导原则(2017年修订版)》(国家食品药品监督管理总局通告2017年第75号)提供产品有效期(货架寿命)的验证资料。不同包装、不同灭菌方式的产品应分别提供验证资料。灭菌验证资料中需要明确灭菌产品的包装材料、包装工艺及方法、加速老化试验或/和实时老化试验报告。加速老化试验中应明确试验温度、湿度、加速老化时间的确定依据;老化试验后需要对包装完整性和包装强度的评价试验,如染色液穿透试验、气泡试验、材料密封强度试验、模拟运输等;若注册申请人提供其他医疗器械产品的灭菌验证资料,则应提供其与本次申报产品在原材料、灭菌方法、灭菌剂量、包装材料、包装工艺、包装方式及其它影响阻菌性能的因素方面具有等同性的证明资料。
6.其他
在新型全膝关节假体产品设计开发过程可能还涉及诸多新技术新方法的应用,如全膝关节假体运动学、关节面接触面积和接触应力分布的研究等,注册申请人可参考附录2中的相关内容,在此不进行强制规定。
(九)产品技术要求的主要性能指标
产品技术要求应按照《医疗器械产品技术要求编写指导原则》(国家食品药品监督管理总局通告2014年第9号)进行编写。
注册产品技术要求应符合相关国家标准、行业标准、国际标准和有关法律、法规的相应要求。在此基础上,注册申请人应根据产品的特点,制定保证产品安全、有效、质量可控的技术要求。膝关节假体技术要求中的性能指标及检验方法的确定是技术要求的主要内容,应根据现行有效的相关国家标准、行业标准、国际标准制定,如YY 0502等,应注明相应标准的编号和年代号。产品技术要求中指标应针对终产品制定,且性能指标不应低于产品适用的强制性国家标准/行业标准,检验方法应优先考虑采用公认的或已颁布的标准检验方法,包括推荐性标准。
对同一注册单元中存在多种型号和/或规格的产品,应明确各型号及各规格之间的所有区别,并附相应图示及数据表格对逐型号规格进行说明。
(十)同一注册单元内注册检验典型性产品确定原则和实例
典型性产品的选择:产品应按不同材质分别选取典型性产品进行注册检测,在同一注册单元内,股骨髁、胫骨衬垫、胫骨托、髌骨衬垫、髌骨托、延长柄、袖套、填充块等应分别检测。
(十一)产品生产制造相关要求
1.详述产品的生产过程,提供生产工艺流程图;
2.明确特殊过程和关键工序,提供特殊过程的确认资料以及关键工序的验证资料(如股骨髁、胫骨衬垫的曲面加工等);
3.明确表面处理工艺(如喷砂、喷丸、等离子喷涂、烧结、气相沉积等);
4.明确生产工艺中涉及的各种加工助剂(如切削液、粘结剂、抛光剂等)质量控制标准;
5.明确产品的清洗过程及清洗验证资料,建议参照ISO 19227,关注产品经末道清洗后加工助剂残留控制情况。
(十二)产品的临床评价要求
注册申请人应在满足注册法规要求的前提下,按照《医疗器械临床评价技术指导原则》(国家食品药品监督管理总局通告2015年第14号)的要求选择合适的临床评价路径提交临床评价资料。
选择通过同品种医疗器械临床试验或临床使用获得的数据进行分析评价,需首先将申报产品与同品种医疗器械进行对比,详述二者的等同性和差异性,分析差异性是否对产品的安全有效性产生不利影响,应通过申报产品自身的数据进行验证和/或确认,如申报产品的非临床研究数据、临床文献数据、临床经验数据,针对差异性在中国境内开展的临床试验的数据,或采用符合要求的境外临床数据,并论证人种差异可能对评价造成的影响。
境外注册申请人如有境外临床试验数据,参照《接受医疗器械境外临床试验数据技术指导原则》(国家食品药品监督管理总局通告2018年第13号)的要求提交相关临床试验资料。
选择国内进行临床试验,则应严格按照《医疗器械临床试验质量管理规范》(国家食品药品监督管理总局、中华人民共和国国家卫生和计生委员会令第25号)进行临床试验,并提交完整的临床试验资料,应当包括临床试验协议、临床试验方案和临床试验报告等。临床试验的设计可参考《医疗器械临床试验设计指导原则》(国家食品药品监督管理总局通告2018年第6号)。开展临床试验研究时,在临床试验方案制定中建议考虑以下因素,包括但不限于:
1.产品预期用途
同一注册单元内的产品,注册申请人应根据预期申报的适用范围不同(初次置换、翻修、肿瘤关节置换),分别对不同类型的产品进行试验。
2.临床试验设计类型
考虑产品为膝关节永久性替代植入物,属于较高风险医疗器械,为了保证试验结果的真实客观性和可比性,建议采用随机对照临床试验。
如果注册申请人采用非随机平行对照研究,在疗效评价时,应考虑基线不均衡而导致无法客观的评价试验结果的可能,注册申请人应提供充分的理由解释结果的客观性和真实性。
如有证据表明同类全膝关节假体在实际临床使用中疗效得到公认,且本次申报产品在设计及生产工艺未发生实质变化,可在临床试验设计中应用单组目标值法,即临床试验不设立对照组。临床试验设计应针对产品与同类产品比较后的差异性进行充分评估。
3.入选、排除标准
受试者应从需要进行全膝关节置换术治疗的一般诊断人群中选出。注册申请人及临床研究机构应根据申报产品的设计特征及其适用范围制定其临床试验的入选/排除/退出标准,不符合所有入选标准或者符合任何一项排除标准的研究对象应被排除。
举例如下:
初次置换骨水泥型全膝关节假体临床试验入选标准:
(1)患者年龄在50-80岁,性别不限。低龄患者应有手术紧迫性需求。
(2)患者骨骼已成熟。
(3)产品适用于退行性骨关节炎、创伤性关节炎或缺血性坏死、炎性关节病等各种终末期膝关节疾病,以及畸形的矫正、部分膝关节假体失败后重建、应用其它技术无法处理的骨折等情形,须行全膝关节假体置换时使用。
(4)受试侧患肢为初次接受全膝关节置换。
(5)术前,受试者或监护人愿意并且能够签署知情同意书。
排除标准:
(1)患者神经肌肉功能不全(例如:麻痹、肌溶解或肌无力)会导致术后膝关节不稳定或步态异常。
(2)患者精神上无能力或者不能理解参与研究的要求。
(3)酗酒者或吸毒者、药物滥用者。
(4)预计依从性差。
(5)肥胖BMI>35。
(6)已知患者对一种或多种植入的材料有过敏史。
(7)膝关节或身体其他部位存在活动性感染病灶。
(8)膝关节周围具有严重的骨质疏松、代谢性骨病、放射性骨病、肿瘤。
(9)妊娠或哺乳期妇女。
(10)体质虚弱或因全身其他疾病不能耐受手术者。
(11)受试者合并的其他疾病限制其参加研究,不能依从随访或影响研究的科学性完整性。
(12)受试者入选前曾参加过其他药物、生物制剂或医疗器械临床研究而未达到主要研究终点时限者。
对于非骨水泥型全膝关节假体、翻修型全膝关节假体、肿瘤型全膝关节假体等产品,应根据产品特点制定入选标准和排除标准。
4.受试者退出标准及退出受试者的处理
退出标准:
(1)受试者撤回知情同意书;
(2)严重违反临床试验方案;
(3)研究者认为不再适合继续进行临床试验者;
(4)在临床试验期间妊娠的妇女;
(5)受试者死亡;
(6)受试者失访;
(7)注册申请人要求终止临床试验。
退出受试者的处理:
(1)所有退出受试者均应保留全部源数据和源文件。当受试者退出后,研究者应采取多种形式如电话、邮件等尽可能与受试者联系,询问理由;
(2)将终止临床试验的时间和原因详细记录在病例报告表上;
(3)因不良事件而脱落者如经随访最终判断与试验器械存在因果关系,必须记录在CRF表中,并通知注册申请人。对因不良事件而终止试验的病人必须随访至不良事件得到解决或稳定。
(4)《医疗器械临床试验质量管理规范》规定的其他相关事宜。
5.临床试验持续时间与窗口期
临床试验的持续时间取决于所有安全性和有效性数据的获得,研究病例至少随访至12个月以上。建议至少在手术前、术后1周内、术后6周、6个月、12个月的膝关节置换假体系统的临床研究数据进行收集,每次随访建议包括患者主诉、体格检查、关节功能评分、安全性评估以及指导患者功能康复等内容。X线片应包括:术前、术后1周内、6个月、12个月各时间点。
6.临床试验评价指标及判定标准
临床试验纳入病例一般应为单侧膝关节置换,如病患需行双侧置换,应在单侧植入至少3个月后,经过评价不会对另外一侧产生影响,方可施行膝关节置换。否则双侧置换病例应选取疗效相对差的一侧进行评价。
(1)主要评价指标
a.主要评价指标:术后12个月评分分数、术前术后12个月评分改变分数、或术后12个月评分“优良率”(即:置换膝关节假体后评分达到优、良的比例。)
b.评分方法:初次置换型、翻修型膝关节可采用KSS评分(下载地址:http://kneesociety.org/the-knee-society-score),KSS评分满分100分,85分以上为优,70-84分为良,60-69分为可,小于60分为差;肿瘤型膝关节可采用MSTS评分(见附录3),该系统满分30分,≥24分定义为优,18-23分定义为良,12-17分定义为中,≤11分定义为差。
(2)次要评价指标
a.X射线平片参数:注册申请人应制定影像学成功的标准并进行评价。同时关注假体周围透亮线的形成(骨水泥质量、异位骨化、假体松动)、骨质溶解、假体位置变化(假体下沉、倾斜、内翻或外翻等)、关节脱位等的发生率。
b.生存率:根据取出或者包括取出膝关节假体的任何一部分的病例来计算假体的生存率。发生1例与产品质量相关的严重不良事件,判定临床试验失败。
c.不良事件发生率。全膝关节置换不良事件的类型包括但不限于:假体松动、感染、关节不稳、对线不良、伤口坏死或愈合不良、异常磨损、假体周围骨折、脱位、关节僵硬、假体断裂、深静脉血栓、心脑血管事件等。
7.对照产品的选择
对照产品应选择目前临床正广泛使用的、疗效已被证实并得到公认的、来源于单一厂家生产的同一系统产品。对照产品的材料、设计、适应证与试验产品具有可比性。注册申请人应提供对照产品的选择依据。
8.样本量的估算
注册申请人应提供样本量足以评价所申报产品安全性和有效性的统计学证据,包括以下内容:对照组与试验组主要评价指标相同试验条件(同样的适应证人群、治疗时间、随访时间等)下的预期疗效、预期的组间差异、显著性水平(α)、把握度(β)、预期失访率、所用到的样本量计算公式、所有的计算过程及使用的统计学软件、引用的参考文献等。
注册申请人应根据产品的性能指标选择对照品,并采用经典的统计学方法及国内外公认的统计学软件计算样本量。
决定样本量的关键因素有:研究类型、主要评价指标、对照组与试验组主要评价指标的预期疗效、非劣效界值或目标值、显著性水平(α)、把握度(β)、预期失访率等。
若进行随机对照非劣效试验,则需明确对照产品预期疗效和临床认可的非劣效界值;若进行单组目标值试验,则需明确试验产品预期疗效和临床认可的目标值。
9.人口统计学和基线特征
(1)人口统计学资料:如性别、年龄、民族、身高、体重等;
(2)临床疗效相关的基线数据:考虑因素包括疾病的诊断、分期、分级及影像学参数、伴随疾病、其他关节问题、风险因素、放射学描述;
(3)合并症:是否有骨质疏松、营养不良(钙、磷、蛋白质、铁)、贫血、激素缺乏(生长激素、甲状旁腺素等)、放射治疗、手术史、糖尿病史、高血压、冠心病、肺功能情况、免疫学疾病等。
10.统计分析方法
应明示具体的统计分析方法以及统计分析软件及版本。
数据分析时应考虑数据的完整性,所有签署知情同意并使用了受试产品的受试者必须纳入分析。数据的剔除或偏倚数据的处理必须有科学依据和详细说明。
临床试验的数据分析应基于不同的分析集,通常包括全分析集(Full Analysis Set,FAS)、符合方案集(Per Protocol Set,PPS)和安全集(Safety Set,SS),研究方案中应明确各分析集的定义。全分析集中脱落病例,其主要研究终点的缺失值的填补方法等应在方案中事先予以说明,并进行不同分析策略的灵敏度分析,以评价缺失数据对研究结果稳定性的影响。
主要研究终点指标的分析应同时在全分析集和符合方案集上进行;安全性指标的分析应基于安全集。
临床试验数据的分析应采用国内外公认的经典统计分析方法。临床试验方案应该明确统计检验的类型、检验假设、判定疗效有临床意义的界值(非劣效界值)等,界值的确定应有依据。
对于主要研究终点,统计结果需采用点估计及相应的95%可信区间进行评价。不能仅将p值作为对主要研究终点进行评价的依据。
对试验期间发生的所有不良事件的种类、严重程度、发生频率及与试验产品的关系将列表描述。
注册申请人应提供基于所有临床试验数据的统计分析报告,以便临床试验组长单位根据此报告撰写临床试验总结报告。
11.其他
已取得产品注册证,且适用范围为初次置换的产品,在新增产品与该注册证产品属于同一注册单元的情况下,增加适用于翻修的产品型号规格及适用范围,可在同时满足如下条件的情况下予以增加:取得注册证满2年、取得注册证后销售的初次置换产品随访数量至少达到100例且产品植入12个月后优良率不低于90%、完成相关功能性试验、备齐相关注册文件。
(十三)产品的不良事件历史记录
应按要求收集、记录、提交产品相关的不良事件记录。不良事件报告应参考通用术语标准,及时、准确上报。全膝关节产品不良事件的类型包括但不限于:假体松动、感染、关节不稳、对线不良、伤口坏死或愈合不良、异常磨损、假体周围骨折、脱位、关节僵硬、假体断裂、深静脉血栓、心脑血管事件等。
(十四)产品说明书和标签要求
1.产品说明书、标签和包装标识应符合《医疗器械说明书和标签管理规定》(国家食品药品监督管理总局令第6号)要求,还应符合相关国家标准、行业标准的要求,例如YY/T 0466.1-2016《医疗器械用于医疗器械标签、标记和提供信息的符号第1部分:通用要求》。
2.产品临床适用范围/适应证、禁忌证、注意事项应依据临床评价或临床试验的结果进行确定。
3.产品有效期、灭菌产品采用的灭菌方法、非灭菌产品推荐采用的灭菌方法等信息应与产品技术报告所述一致。
4.说明书的警示中注明MRI内容,提示其存在的风险。
三、参考文献
1.《医疗器械监督管理条例》(中华人民共和国国务院令第650号)
2.《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号)
3.《医疗器械临床试验质量管理规范》(国家食品药品监督管理总局、中华人民共和国国家卫生和计生委员会令第25号)
4.《医疗器械说明书和标签管理规定》(国家食品药品监督管理总局令第6号)
5.《医疗器械临床评价技术指导原则》(国家食品药品监督管理总局通告2015年第14号)
6.《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》(国家食品药品监督管理总局公告2014年第43号)
7. YY/T 0316-2016《医疗器械风险管理对医疗器械的应用》
8.《无源植入性医疗器械产品注册申报资料指导原则》(食药监办械函〔2009〕519号)
9.《关于发布无源植入性医疗器械货架有效期注册申报资料指导原则(2017年修订版)的通告》(国家食品药品监督管理总局通告2017年第75号)
10.《医疗器械临床试验设计指导原则》(国家食品药品监督管理总局通告2018年第6号)
11.《医疗器械注册单元划分指导原则》(国家食品药品监督管理总局通告2017年第187号)
12. Guidance for Industry and FDA Class II Special Controls Guidance Document: Knee Joint Patellofemorotibial and Femorotibial Metal/Polymer Porous-Coated Uncemented Prostheses
13. Guidance for Industry and FDA Class II Special Controls Guidance Document: FDA Guidance: Characterization of Ultrahigh Molecular Weight Polyethylene (UHMWPE)Used in Orthopedic Devices
14.《无源植入性骨、关节及口腔硬组织个性化增材制造医疗器械注册技术审查指导原则》(国家药品监督管理局通告2019年第70号)
四、编写单位
本指导原则由国家药品监督管理局医疗器械技术审评中心编写并负责解释。
附录1
全膝关节假体产品风险管理资料要求
一、总要求
注册申请人应提供产品上市前对其风险管理活动进行全面评审所形成的风险管理报告以及相关的产品风险管理资料。该风险管理报告应说明:
-风险管理计划已被适当地实施;
-综合剩余风险是可接受的;
-已有恰当的方法获得与本产品相关的包括出厂后流通与临床应用的信息。
除此之外,风险管理报告还应扼要说明:
-在产品研制的初期阶段,对风险管理活动的策划,和所形成的风险管理计划;
-说明已识别了产品的有关可能的危险(源),并对其危险(源)产生的风险进行了估计和评价;
-在降低风险措施方面,考虑了相关安全标准和相关产品标准,并有针对性地实施了降低风险的技术和管理方面的措施;
-通过产品性能、成分等测试、生产工艺检验、相关文件的审查、试生产等活动对风险控制措施的有效性实施验证;
-注册申请人应对产品的安全性作出承诺。
二、风险管理报告及风险管理资料内容
(一)职责权限
注册申请人应明确参与风险管理活动的成员,包括风险分析人员、风险评价人员、风险控制措施制定人员及验证人员、风险管理过程评审人员以及风险管理报告的编制及审批人员,应列出其姓名、职务及责任范围。其成员应具有与风险管理任务相适应的知识和经验。
(二)产品描述
-通过照片、结构图和文字等形式说明产品的各型号、各组件、以及各关键部位;
-产品各组件的材料牌号;
-产品工艺流程。
(三)产品适用标准
(四)产品的安全特征判定
注册申请人可参照YY/T 0316-2016《医疗器械风险管理对医疗器械的应用》附录C提示的问题,对照产品的实际情况简明扼要的描述产品安全特征。产品如存在附录C提示以外的可能影响安全性的特征时,也应做出说明。最终形成一份《产品安全特征清单》。
(五)产品的可能危险(源)判定
注册申请人应在《产品安全特征清单》的基础上,系统地识别产品在正常和故障两种条件下的可预见的危险(源)。并对危险(源)的成因及后果进行分析,即说明危险(源)、可预见事件序列、危险情况和可能发生的伤害之间的关系。形成一份产品可预见的危险(源)及危险(源)分析清单。
下表为膝关节假体常见危险(源)举例,注册申请人应考虑不同型号、不同材料等危险(源),并补充危险(源)清单之中:
|
可预见的危险(源) |
危险(源)举例及形成因素 |
|
生物相容性方面的危险(源) |
膝关节假体产品材料(包括如表面涂层处理后)生物不相容,如:毒性、致热源等 |
|
化学危险(源) |
化学成分不符 |
|
金属腐蚀 |
|
|
产品清洗后的酸碱性 |
|
|
加工过程中使用的材料(清洁剂等)未能有效清除 |
|
|
环氧乙烷灭菌后的环氧乙烷残留 |
|
|
生物学方面的危险(源) |
产品未能有效灭菌 |
|
产品包装未能有效阻菌 |
|
|
交叉感染 |
|
|
涂层剥落、过度磨损造成的局部组织反应,如:磨屑、金属离子带来的骨溶解、过敏反应等 |
|
|
机械能方面的危险(源) |
疲劳破坏造成的部件断裂等 |
|
过度磨损 |
|
|
配合尺寸不符,松动、位移、脱位、微动腐蚀等 |
|
|
产品表面缺陷 |
|
|
产品使用错误的危险(源) |
操作者应经过何种培训 |
|
手术时,选择了不适当的规格型号的产品 |
|
|
说明书、标签方面的危险(源) |
说明书不完整,不符合要求 |
|
标签内容缺失、错误 |
|
|
可追溯标记 |
|
|
…… |
|
(六)对危险(源)清单中每一危险情况下的风险进行风险估计和风险评价
注册申请人应明确风险可接受准则,并对伤害发生的概率和伤害的严重程度予以明确定义;产品国家标准、行业标准、国际标准中如涉及了相关风险的可接受准则,该准则应作为注册申请人所确定的风险可接受准则之一,除非有证据证实其特定风险的可接受准则不必符合相关标准。注册申请人应依据风险可接受准则对危险(源)清单中每一危险(源)处境下的风险进行风险估计和风险评价。
风险评价的结果可以记入《风险管理文档》中。
(七)降低风险的控制措施
注册申请人应对经风险评价后不可接受的、或考虑可进一步采取措施降低的风险实施降低风险的控制措施。在制定降低风险的控制措施方案时,应充分考虑产品国家标准、行业标准、国际标准中有关降低风险的措施。应确保降低风险的控制措施在研制初期得到有效的输入,并应对措施的有效性实施验证。
注册申请人应对采取降低风险的控制措施后的剩余风险以及是否会引发新的风险进行评价。
以上降低风险的控制措施、控制措施的验证、剩余风险评价等信息可以记入《风险评价、风险控制措施以及剩余风险评价汇总表》中。
(八)结论
注册申请人应对综合剩余风险是否可接受给出结论性意见,并对已有恰当的方法获得与本产品相关的包括出厂后流通与临床应用的信息进行阐述并作出承诺。
风险管理报告应由注册申请人的最高管理者(法人)或其授权的代表签字批准。
附录2
资料性附录
本附录内容,注册申请人在产品研发时依据产品特征予以关注。
一、关节面接触面积和接触应力分布研究
全膝关节假体关节面的磨损与接触应力、接触面积相关,通过测试接触应力、接触面积有助于预测假体关节面的磨损性能[1-2],从而为假体关节面设计改进以及磨损试验最差情况的选择提供依据。
对于胫股关节面,建议在屈曲角为0°、15°、30°、60°、90°和最大屈曲角度时分析或者测试接触应力和接触面积,并在90°和最大屈曲角处时带有0°旋转和15°内旋/外旋进行试验,对于内旋外旋活动度具有一定限制的假体,如髁限制型假体,内旋外旋角度可能达不到15°,需根据假体轴向旋转活动度选择适宜的角度进行试验。对于活动平台假体,应对所有的接触面(股骨-胫骨衬垫、胫骨衬垫-胫骨托)进行接触面积和接触应力的测试。上述接触应力和接触面积分布的测试可参照ASTM F2083的方法进行分析或试验。
采用体外试验方法测试胫股关节面接触应力和接触面积时,将股骨髁和胫骨部件固定在与假体匹配的模拟骨或金属夹具上,胫骨部件夹具可允许具有一定的前后方向、轴向旋转和内翻外翻自由度,当施加载荷时,胫骨部件可以微调到达平衡位置。将夹具固定在力学试验机上,使加载轴线通过膝关节假体股骨髁间窝中心或者模拟膝关节假体生产企业推荐的力线对线、倾角(如胫骨部件后倾角)要求。对于电子压力测试装置(如K-scan),需要在试验前按照设备要求进行传感器校准和计量,对于压敏薄膜,需要根据试验选择的载荷水平选取相适应的压力级别薄膜进行测试。将电子压力测试装置的传感器或压敏薄膜置于关节面之间,预施加载荷观察接触区域是否完全在传感器或压敏薄膜边界内,在不同的屈曲角度下施加不同的载荷[3-6],记录相应的关节面的接触应力和接触面积。上述试验过程仅描述了屈曲角度和载荷,为模拟体内股骨髁和胫骨部件的相对位置,可进一步考虑在不同屈曲角度下股骨外旋(或胫骨内旋)、前后位移和内翻外翻角度对接触应力和接触面积的影响,设置相应的试验条件测试接触应力和接触面积分布。此外,除电子压力测试装置和压敏薄膜外,也可采用超声测量装置测试膝关节假体关节面的接触应力和接触面积分布[7]。
对于髌股关节面,建议在屈曲角为15°、45°、90°和最大屈曲角度时在相应的载荷下测试髌股关节面的接触应力和接触面积,可参照ASTM F1672的方法或文献[8-13]中描述的方法进行试验或有限元分析,需描述具体的测试方法(包括股骨与髌骨的相对位置、加载载荷、固定夹具等)、试验设备和材料、测试结果。
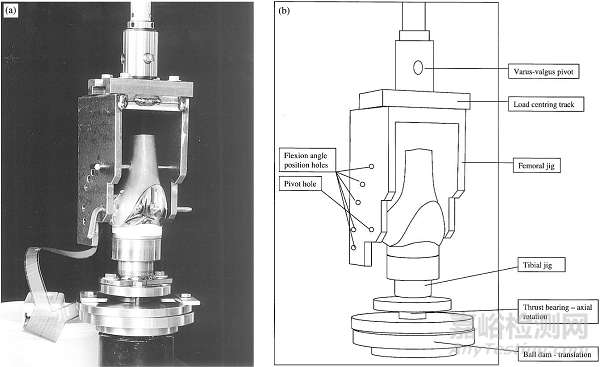
图1 接触应力和接触面积试验示意图
参考文献
[1] Rostoker W, Galante J. O. Contact pressure dependence of wear rates of ultra high molecular weight polyethylene. J Biomed Mater Res, 1979, 13: 957.
[2] Abdellatif Abdelgaied, Claire L Brockett, Feng Liu, Louise M Jennings, Zhongmin Jin and John Fisher. The effect of insert conformity and material on total knee replacement wear. Proceedings of the Institution of Mechanical Engineers, Part H: Journal of Engineering in Medicine, 2014, 228: 98-106.
[3] Szivek, J. A., Cutignola, L., Volz, R. G. Tibiofemoral Contact Stress and Stress Distribution Evaluation of Total Knee Arthroplasties. J. Arthroplasty, Vol 10, No. 4, 1995, pp. 480–491.
[4] Harris, M. L., Morberg, P., Bruce, W. J. M., Walsh, W. R. An Improved Method for Measuring Tibiofemoral Contact Areas in Total Knee Arthroplasty: A Comparison of K-Scan Sensor and Fuji Film. Journal of Biomechanics, Vol 32, 1999, pp. 951-958.
[5] DeMarco, A. L., Rust, D. A., Bachus, K. N. Measuring Contact Pressure and Contact Area in Orthopedic Applications: Fuji Film vs Tekscan. Orthopedic Research Society, March 12-15, 2000, Orlando, FL, p. 518.
[6] Otto, J. K., Brown, T. D., Heiner, A. D., Callaghan, J. J. Heredity Integral Drift Compensation in Piezoresistive Contact Stress Sensors. Orthopedic Research Society, February 1-4, 1999, Anaheim, CA, p. 957.
[7] Zdero R., Fenton P. V., Rudan J., Bryant J. T. Fuji film and ultrasound measurement of total knee arthroplasty contact areas. The Journal of Arthroplasty, 2001, 16(3): 367-375.
[8] McNamara, J. L., Collier, J.P., Mayor, M.B., and Jensen, R. E. A Comparison of Contact Pressures in Tibial and Patellar Total Knee Components Before and After Service In-Vivo. Clin. Orthop. Rel. Res., No. 299, 1994, pp. 104–113.
[9] Morra, E. A., and Greenwald, A. Seth. Patello-Femoral Replacement Polymer Stress During Daily Activities: A Finite Element Study. AAOS 2006, Orthopaedic Research Laboratories, Cleveland, OH: http://orl-inc.com.
[10] Reilly, D. T., and Martens, M. Experimental Analysis of the Quadriceps Muscle Force and Patello-Femoral Joint Reaction Force for Various Activities. Acta Orthop Scand, Vol 43, 1972, pp. 126–137.
[11] Mathews, L. S., Sonstegard, D. A., and Henke, J. A., Load Bearing Characteristics of the Patello-Femoral Joint. Acta Orthop Scand, Vol 48, 1977, pp. 511–516.
[12] Sharma, A., Leszko, F., Komistek, R. D., Scudery, G. R., Cates, H. E., and Liu, F. In Vivo Patellafemoral Forces in High Flexion Total Knee Arthroplasty. Journal of Biomechanics, Vol 41, 2008, pp. 642–648.
[13] Mason, J., Leszko, F., Johnson, T., and Komistek, R. D. Patellofemoral Joint Forces. Journal of Biomechanics, Vol 41, 2008, pp.2337–2348.
二、全膝关节假体运动学评价
(一)膝关节运动学评价的意义
人工全膝关节置换术(TKA)是治疗晚期骨性关节炎常用的手段,但TKA术后约17%的病人对手术效果不满意[1]。导致病人不满意的因素很多,但ROM活动度受限(10%)以及膝前痛(5%-10%)是引起术后病人不满意的最主要原因[2]。在前后方向股骨髁的向前滑移可以导致接触点前移,并由于关节碰撞引起疼痛。股四头肌更短的力臂还会导致髌骨高应力和髌前痛[3]。除股骨髁后滚不足外,全膝关节置换术(TKA)后病人的运动学普遍存在旋转角度变小的现象[4-6],这将影响髌骨运动轨迹、关节稳定性,还会使病人本体感觉下降[7]。髌骨假体和股骨髁滑车的设计不佳会造成髌股关节面不稳、髌骨运动轨迹不良和增加髌骨脱位风险[8-10],髌骨假体的材料性能、髌骨与股骨髁的相互作用力、关节面设计和运动范围会对髌骨假体的磨损性能产生影响[11-13]。综上所述,在全膝关节假体设计开发中,评估胫股关节面和髌股关节面的临床前运动学有助于改进假体和手术工具设计,从而改善TKA术后结果和患者满意度。
(二)全膝关节假体运动学评价的目标
对全膝关节假体进行运动学评价的目标是通过分析或试验方法模拟全膝关节假体在植入人体中的受力条件预测假体的胫股关节面和髌股关节面的运动学表现,尽可能避免假体设计和手术工具设计等因素导致的假体临床使用中的运动学风险,如异常前移、轴向旋转量不足、中段屈曲不稳、髌骨假体不稳、脱位和磨损等,从而改进全膝关节假体的临床表现,提高患者满意率。
(三)运动学评价指标
建议主要评价以下运动学指标:
1.异常前移量:评估屈曲-伸展过程股骨髁内外侧髁与胫骨衬垫的在前后方向的相对位移,尽量模拟膝关节的股骨后滚。
2.轴向旋转量:评估屈曲-伸展过程股骨髁相对于胫骨衬垫的外旋和内旋角度,尽量模拟膝关节的股骨外旋和内旋。
3.运动范围:在模拟膝关节假体生产企业推荐的力线对线要求下,评估假体屈曲角度范围能否到达设计预期。
4.凸轮-立柱接触:凸轮-立柱的接触位置与屈曲角度的关系是否与设计预期相符。
5.特殊设计假体的旋转中心是否达到了设计预期。
6.髌骨假体运动轨迹是否达到设计预期,评价髌骨不稳、脱位以及髌股关节面磨损的风险。
(四)运动学评价方法建议
采用有效的仿真[14-19]或体外试验方法[20-24]均可。
仿真方法应确保模型是经过严格验证且结果准确的,方法是合理的。可采用ISO 14243-1 或ASTM F3141或ISO 14243-5或其他证明有效可靠的力控制输入条件进行动力学计算,或采用经过验证的骨肌多体动力学模型计算。
体外试验方法应确保试验方法(包括曲线控制、假体安装位置准确、定位可靠等)的准确性和合理性,公差控制在允许范围。可采用ISO 14243-1 或ASTM F3141或ISO 14243-5或其他证明有效可靠的力控制输入条件进行运动学试验(需提供输入曲线来源)。对于评价全膝关节假体的髌股关节面磨损风险时,可参照ISO 14243-5标准方法进行试验。
根据假体设计特性,可评价步态、深蹲或坐起、上下楼等其他步态的运动学表现。
运动学仿真或试验结果报告需包含完备的试验方法、过程描述、参数设置、运动学评价结果(同类产品对比)等。
基于持续改善假体临床表现和患者满意度的追求,除上述临床前全膝关节假体运动学评价方法外,也可采用精确的体内运动学分析手段对全膝关节假体的运动学进行研究。目前,常用的体内运动学分析手段包括体内透视分析(in vivo fluoroscopic analyses)[25-28]、运动捕捉分析(noninvasive analyses using motion tracking surface markers)[29]、伦琴立体摄影测量分析(roentgen stereophotogrammetric analysis,RSA)[30]、准静态MRI分析(quasi-dynamic magnetic resonance imaging testing)[31-32]。
参考文献
[1] Daniilidis K., Skwara A., Vieth V., et al. Highly conforming polyethylene inlays reduce the in vivo variability of knee joint kinematics after total knee arthroplasty[J]. Knee, 2012, 19(4): 260-265.
[2] McCalden R.W., MacDonald S.J., Charron K.D., et al. The role of polyethylene design on postoperative TKA flexion: an analysis of 1534 cases[J]. Clin Orthop Relat Res, 2012, 468(1): 108-114.
[3] Van Stralen R.A., Heesterbeek P.J., Wymenga A.B. Different femorotibial contact points between fixed- and mobile-bearing TKAs do not show clinical impact[J]. Knee Surg Sports Traumatol Arthrosc, 2015, 23(11): 3368-3374.
[4] Louisia S., Siebold R., Canty J., Bartlett R.J. Assessment of posterior stability in total knee replacement by stress radiographs: prospective comparison of two different types of mobile bearing implants[J]. Knee Surg Sports Traumatol Arthrosc, 2005, 13(6): 476-482.
[5] Minoda Y., Ikebuchi M., Mizokawa S., et al. Mobile-bearing TKA improved the anteroposterior joint stability in mid-flexion range comparing to fixed-bearing TKA[J]. Arch Orthop Trauma Surg, 2016, 136(11): 1601-1606.
[6] W. NORMAN SCOTT, MD, FACS. Insall & Scott Surgery of the Knee, Sixth Edition. 2018, P379-379.
[7] Walker P.S., Sussman-Fort J.M., Yildirim G., et al. Design features of total knees for achieving normal knee motion characteristics[J]. J Arthroplasty, 2009, 24(3): 475-483.
[8] Petersilge WJ, Oishi CS, Kaufman KR, et al. The effect of trochlea design on patellofemoral shear and compressive forces in total knee arthroplasty. Clin Orthop, 1994, 309: 124-130.
[9] Theiss SM, Kitziger KJ, Lotke PS, et al. Component design affecting patellofemoral complications after total knee replacement. Clin Orthop, 1996, 326: 183-187.
[10] Yoshii I, Whiteside LA, Anouchi YS. The effect of patella button placement and femoral design on patellar tracking in total knee arthroplasty. Clin Orthop, 1992, 275: 211-219.
[11] Conditt MA, Noble PC, Allen B, et al. Surface damage of patellar components used in total knee arthroplasty. J Bone Joint Surg Am, 2005, 87: 1265-1271.
[12] De Swart RJ, Stulberg BN, Gaisser DM, et al. Wear characteristics of all-polyethylene patellar components: a retrieval analysis. Trans Orthop Res Soc, 1989, 14: 367.
[13] Hsu H-P, Walker PS. Wear and deformation of patellar components in total knee arthroplasty. Clin Orthop, 1989, 246: 260-265.
[14] Mark A. Baldwin, Chadd W. Clary, Clare K. Fitzpatrick, James S.Deacy, Lorin P. Maletsky, Paul J. Rullkoetter. Dynamic finite element knee simulation for evaluation of knee replacement mechanics. Journal of Biomechanics, 2012, 45 (3): 474-483.
[15] Fabio Catani, Bernardo Innocenti, Claudio Belvedere, Luc Labey, Andrea Ensini, Alberto Leardini. Articular Contact Estimation in TKA Using In Vivo Kinematics and Finite Element Analysis. Clin Orthop Relat Res, 2010, 468:19–28.
[16] Jason P. Halloran, Anthony J. Petrella, Paul J. Rullkoetter. Explicit finite element modeling of total knee replacement mechanics. Journal of Biomechanics, 2005, 38: 323–331.
[17] Xiao-Hong Wang, Da-Yong Song, Xiang Dong, Toru Suguro, Cheng-Kung Cheng. Motion type and knee articular conformity influenced mid-flexion stability of a single radius knee prosthesis [J]. Knee Surgery Sports Traumatology Arthroscopy, 27:1595-1603,2019.
[18] Clare K. Fitzpatrick, Chadd W. Clary, Adam J. Cyr, Lorin P. Maletsky, Paul J. Rullkoetter. Mechanics of Post-Cam Engagement during Simulated Dynamic Activity. J Orthop Res, 2013, 31:1438–1446.
[19] Oliver S. Schindler. Basic kinematics and biomechanics of the patellofemoral joint Part 2: The patella in total knee arthroplasty. Acta Orthop. Belg., 2012, 78: 11-29.
[20] Lee K.M., Guo J. Kinematic and dynamic analysis of an anatomically based knee joint[J]. J Biomech, 2010, 43: 7231-7236.
[21] Hani Haider, Peter Walker, John DesJardins, Gordon Blunn. Effects of Patient and Surgical Alignment Variables on Kinematics in TKR Simulation Under Force-Control. Journal of ASTM International, 2006, 3(10): 3-16.
[22] Li G., Most E., Sultan P.G., et al. Knee kinematics with a high-flexion posterior stabilized total knee prosthesis: an in vitro robotic experimental investigation[J]. J Bone Joint Surg Am, 2004, 86-A(8): 1721-1729.
[23] Peter S. Walker, Hani Haider. Characterizing the Motion of Total Knee Replacements in Laboratory Tests. Clinical Orthopaedics and Related Research, 2003, Number 410, pp. 54-68.
[24] Kartik M. Varadarajan, Rubash E. Harry, Todd Johnson, Guoan Li. Can in vitro systems capture the characteristic differences between the flexion–extension kinematics of the healthy and TKA knee? Medical Engineering & Physics, 2009, 31: 899-906.
[25] Marc R. Angerame, David C. Holst, Jason M. Jennings, Richard D. Komistek, Douglas A. Dennis. Total Knee Arthroplasty Kinematics. The Journal of Arthroplasty, 2019, 34(10): 2502-2510.
[26] Ranawat CS, Komistek RD, Rodriguez JA, et al. In vivo kinematics for fixed and mobile-bearing posterior stabilized knee prostheses. Clin Orthop Relat Res, 2004, 418: 184-190.
[27] Victor J, Mueller JKP, Komistek RD, et al. In vivo kinematics after a cruciate substituting total knee TKA. Clin Orthop Relat Res, 2010, 468: 807-814.
[28] Trevor F. Grieco, Adrija Sharma, Garett M. Dessinger, Harold E. Cates, Richard D. Komistek. In Vivo Kinematic Comparison of a Bicruciate Stabilized Total Knee Arthroplasty and the Normal Knee Using Fluoroscopy. The Journal of Arthroplasty, 2018, 33(2): 565-571.
[29] Schulz BW, Kimmel WL. Can hip and knee kinematics be improved by eliminating thigh markers? Clin Biomech (Bristol, Avon), 2010, 25: 687-692.
[30] Benoit DL, Ramsey DK, Lamontagne M, et al. Effect of skin movement artifact on knee kinematics during gait and cutting motions measured in vivo. Gait Posture, 2006, 24: 152164.
[31] Johal P, Williams A, Wragg P, et al. Tibio-femoral movement in the living knee. A study of weight bearing and non-weight bearing knee kinematics using “interventional” MRI. J Biomech, 2005, 38: 269-276.
[32] H. Iwaki, V. Pinskerova, M. A. R. Freeman. Tibiofemoral movement 1: the shapes and relative movements of the femur and tibia in the unloaded cadaver knee. J Bone Joint Surg [Br], 2000, 82-B: 1189-95.
附录3
MSTS评分
MSTS(The Musculoskeletal Tumor Society,世界骨与软组织肿瘤协会)骨肿瘤保肢术后肢体功能评分。
一、肢体疼痛
以口头询问和查看病历的方式进行,了解病人受访当时的疼痛感受、近一个月疼痛影响活动和止疼用药情况。
|
评分 |
状态 |
参考 |
|
5 |
无疼痛 |
不用任何止疼药物。 |
|
4 |
轻度痛 |
有轻微疼痛感,不用药物止疼,或偶用非麻醉镇痛药物。 |
|
3 |
中轻痛 |
疼痛,不致残,应用非麻醉镇痛药物,不用麻醉剂。 |
|
2 |
中度痛 |
有较强烈疼痛感,持续大量使用非麻醉镇痛药物或偶用麻醉剂。 |
|
1 |
中重痛 |
强烈疼痛,疼痛致残,间断性活动障碍,间断使用麻醉剂。 |
|
0 |
严重痛 |
难忍疼痛,疼痛致残,持续性活动障碍,持续使用麻醉剂。 |
二、活动功能
活动功能指患者因活动受限使病前的职业功能丧失的程度,但应考虑职业活动量特征,一般情况下职业活动量和肢体功能应用程度应小于娱乐中的。评价以询问和活动观察为主。
|
评分 |
状态 |
参考 |
|---|---|---|
|
5 |
无限制 |
无活动受限,娱乐和职业与病前一样。 |
|
4 |
轻度限制 |
娱乐和职业与病前一样,但长时间娱乐有不舒服感或力不从心。 |
|
3 |
娱乐限制 |
小部分功能残疾,部分活动功能受限,娱乐活动受到限制,只能选择性或短时间参与,职业轻微影响。 |
|
2 |
中度功能受限 |
一半功能残疾,活动功能受限,职业受到明显影响,如工作时间缩短,工作量下降或效率降低,或更换了病前职业。 |
|
1 |
部分职业限制 |
大部分功能残疾,活动功能明显受限,职业受到很大限制,已不能从事正常活动的职业。 |
|
0 |
所有职业受限 |
完全残疾,活动功能完全受限,不能从事任何职业。 |
三、自我感受
询问病人自我感受,主要是满意度和情感接受程度。
|
评分 |
状态 |
参考 |
|
5 |
非常满意 |
非常热心接受手术,有感激之情,并将此手术介绍给其他人。 |
|
4 |
很满意 |
热心接受手术。 |
|
3 |
满意 |
还可以,假设重新选择,还选择接受该手术。 |
|
2 |
基本满意 |
凑合吧。 |
|
1 |
接受 |
不太满意,但是再次选择的话,勉强可以接受同样的手术。 |
|
0 |
厌恶 |
后悔选择这种手术,宁可去截肢。 |
四、支具使用
调查病人支具使用情况,本研究中需在随访现场准备拐棍和拐杖实际考察,中间分值的病人可在上下两种支撑状态下测试,选择更符合生活情况的一种状态,并录像记录。
|
评分 |
状态 |
参考 |
|---|---|---|
|
5 |
不用 |
不用任何支具。 |
|
4 |
偶尔用支撑 |
偶尔或累了用拐棍(手拄)。 |
|
3 |
经常用支撑 |
大多数时候要拄拐棍。 |
|
2 |
偶尔用拐杖 |
偶尔用拐杖(腋下支撑)。 |
|
1 |
一个拐杖 |
大多数时候要用拐杖,不用拐杖勉强只能少量挪动步伐或单腿跳。 |
|
0 |
两个拐杖 |
若要行走,必须使用两个拐杖,否则不能行走。 |
五、行走能力
不用支具,在室内往返约5米距离测试,室外往返25米距离测试,并录像记录。
|
评分 |
状态 |
参考 |
|
5 |
无限制 |
行走如病前。 |
|
4 |
偶有限制 |
限制较少,外出不受影响。 |
|
3 |
有限制 |
可以外出,但行走能力明显降低,不宜行走过远,如1站地。 |
|
2 |
明显受限 |
不宜外出行走,比较危险。 |
|
1 |
仅能室内行走 |
只能在扶助固定物情况下挪步,无法外出。 |
|
0 |
不能单独行走 |
要位移只能做轮椅或完全依靠外力。 |
六、步态改变
在行走能力测试的同时观察步态变化,并录像记录。
|
评分 |
状态 |
参考 |
|
5 |
正常 |
无改变,不需要掩饰。 |
|
4 |
略微有变化 |
不易察觉,步态不会令人感到诧异。 |
|
3 |
轻度变化 |
步态轻度外观改变,需要掩饰。 |
|
2 |
中度变化 |
步态不正常,需要努力掩饰。 |
|
1 |
明显变化 |
步态明显不正常,一部分功能缺失。 |
|
0 |
严重残疾 |
步态严重障碍,严重功能缺失。 |

来源:国家药监局


