您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2020-02-27 16:57
田霖 tianlin@nifdc.org.cn, 杨会英 , 孙会敏 通信作者sunhm@126.com
中国食品药品检定研究院, 北京 100050
收稿日期:2019-06-25
作者简介:田霖, 博士, 助理研究员, 研究方向:劳动卫生与环境卫生学, Tel:(010)67095124, E-mail:tianlin@nifdc.org.cn
通信作者:孙会敏, Tel:(010)67095721, E-mail:sunhm@126.com
摘要:
目的: 对比分析国内外药包材洁净环境现行相关法规和标准,找出目前我国药包材洁净环境存在的问题和需要改善的方面。方法: 通过比较药包材洁净环境相关法规和标准的异同,发现我国法规和标准有待更新和提高之处,并参照行业现状,指出药包材洁净环境现存问题。结果: 我国GMP与欧盟GMP总体保持一致,但其他的标准在一些参数的测试方法和标准上仍存在空白,我国药包材生产企业在开展日常监控和参数验证等工作中尚存缺陷。结论: 我国药包材洁净环境相关法规和标准已经与国际接轨,一些条款仍需更新和完善。目前我国的药包材洁净环境仍存在一些问题,需要进一步推进相关法规和标准的执行,促进洁净环境整体质量的提升。
关键词:药包材 洁净环境 法规 标准
"药包材"即直接接触药品的包装材料和容器[1]。作为药品的一部分, 药包材本身的质量、安全性、使用性能以及药包材与药物之间的相容性对药品质量有着十分重要的影响[2-3]。药包材存在的问题, 可能会给药物制剂带来微生物污染、可见异物、不溶性微粒超标等安全隐患。药包材生产环境是决定药包材整体质量的一个关键因素。
洁净室是指对空气悬浮粒子及微生物浓度受控的房间或区域, 它的建筑结构、装备和使用应具有减少室内诱入、产生及滞留污染源的功能。室内其他有关参数如温度、湿度、压力等按要求进行控制[4]。药品生产的洁净环境是生产质量保证的一个关键和基础, 国家药品监督管理局已经发布的关联审评审批技术要求中, 对包材辅料的生产环境提出了明确的要求, 药用包材辅料的生产环境应与药品制剂生产环境相匹配[5-7]。药包材生产的洁净环境同样是药品整体质量的一个关键组成部分。
由于药包材生产洁净环境的重要性, 多个国家和组织都针对药包材洁净环境发布了法规和相应标准, 目前国内外现有的药包材洁净环境标准见表 1。我国最早的相关规定是2004年原国家食品药品监督管理局公布的《直接接触药品的包装材料和容器管理办法》(局令第13号)[1]。2010年, 国家发布了《洁净室施工及验收规范》(GB 50591-2010)[8], 明确了洁净室施工和验收相关标准要求。另有《医药工业洁净室(区)悬浮粒子的测试方法》(GB/T 16292-2010)、《医药工业洁净室(区)浮游菌的测试方法》(GB/T 16293- 2010)和《医药工业洁净室(区)沉降菌的测试方法》(GB/T 16294-2010), 分别对洁净环境中悬浮粒子、浮游菌和沉降菌的测试方法作了具体规定[9-11]。《药品生产质量管理规范》[12](GMP)及其《附录1无菌药品》于2010年进行了修订, 内容上基本实现与国际先进法规和标准的接轨。2015年, 中国食品药品检定研究院发布了《国家药包材标准》, 其中有《药品包装材料生产厂房洁净室(区)的测试方法》(YBB00412004-2015)[4], 对药包材生产厂房洁净室各项目的具体测试方法进行了规定。2018年, 我国发布了《洁净室及相关受控环境检测技术分析与应用》(GB36066- 2018)[13], 对洁净室及相关受控环境的检测要求、基本仪器配置和技术要求进行了规定。欧盟、美国、日本和澳大利亚等国也有各自的GMP出台[14-20], 国际药品认证合作组织(Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme, PIC/S)GMP[15]是目前全球公认最严谨的制药规范, 在2018年6月发布了其修订后的最新版本。国际标准化组织(International Organization for Standardization, ISO)在《洁净室及相关受控环境》(ISO 14644 2005)[21-23]和《生物污染控制总则》(ISO 14698 2003)[24-25]中对洁净环境部分参数的测试方法也有体现。
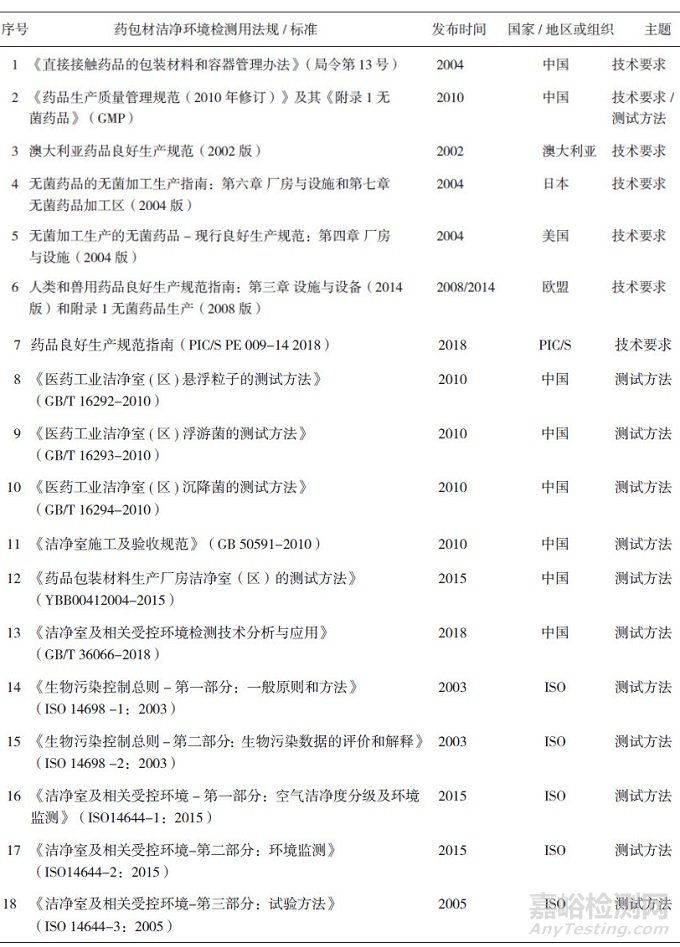
表 1 国内外现有药包材洁净环境检测用法规/标准
不同法规和标准在洁净区域的检测项目、检测方法以及各参数的标准规定等方面有所不同, 现拟从这些方面将现行法规和标准进行比较, 以便综合、全面地理解药包材洁净环境的相关要求。
1 检测项目的异同
国内外的药包材洁净环境标准对于洁净环境的基本检测项目均有所涉及, 中国与其他国家、地区或组织洁净环境检测项目的异同见表 2。其中, 我国2010年版GMP与欧盟GMP在检测项目上要求相同, 检测项目比较齐全, 均需要检测温湿度、压差、悬浮粒子、浮游菌、沉降菌等共计13个项目。GB 50591-2010中除了表面接触菌没有规定外, 其余检测项目均有所涉及。YBB00412004-2015中没有涉及送风量、高效过滤器完整性、自净时间和表面接触菌的测试, 而局令13号由于形成时间比较早, 除上述四项不涉及外, 也没有规定气流流型相关的测试。
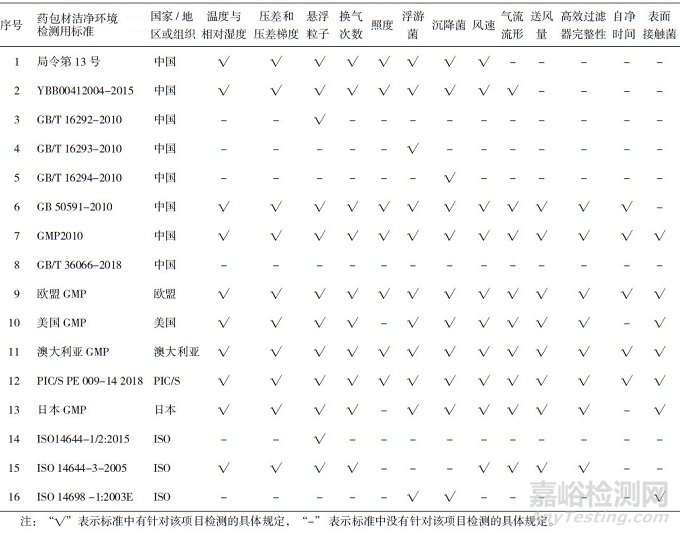
表 2 中国与其他国家、地区或组织洁净环境检测项目对比
2 检测方法的异同
在检测方法上, 各法规和标准对于温度和相对湿度、压差、换气次数、照度、高效过滤器完整性、气流流型、自净时间和风速的方法描述基本一致。我国GMP、药包材标准以及GB/T16292/3, 和ISO 14698 -1:2003相比, 在浮游菌和沉降菌检测方法上的表述没有明显差异。国内外的GMP都明确需要对浮游菌进行动态监测, 根据空气净化系统确认的结果和风险评估来确定采样点的位置, 并且应当制定适当的纠偏警戒限度和纠偏限度, 操作规程中应当详细说明结果超标时需采取的纠偏措施。
对于悬浮粒子的测试, 局令第13号中没有具体描述, 仅提到在静态环境进行。国内外GMP都明确要求对悬浮粒子进行动态监测, 根据空气净化系统确认的结果和风险评估来确定取样点的位置, 并应制定适当的纠偏警戒限度和纠偏限度, 操作规程中应当详细说明结果超标时需采取的纠偏措施。在测试方法上, 我国GMP、GB/T 16292-2010与欧盟GMP要求一致。ISO14644的2015版本相较于1999版, 删除了95%UCL的计算, 取消了计算房间面积的平方根作为最少采样点数的方法, 而用表格法和补充公式, 且对于ISO 5级的5μm微粒不作规定, 目前国内还没有相应标准同步更新。
3 检测参数限度的异同
空气洁净度是洁净环境检测中的一个比较关键的指标, 各标准对于悬浮粒子检测标准的规定有所不同。
PIC/S、日本、欧盟和我国的GMP对于悬浮粒子检测标准的表述一致, 见表 3。美国GMP对悬浮粒子的标准有详细规定:洁净区域控制参数应当得到验证期间获得的微生物和尘埃粒子数据支持, 洁净室的首次验证包括对空态、静态条件下空气质量的评价; 区域验证和分级中, 要将大部分重点放在动态条件下产生的数据。不同洁净度级别悬浮粒子的标准见表 4。
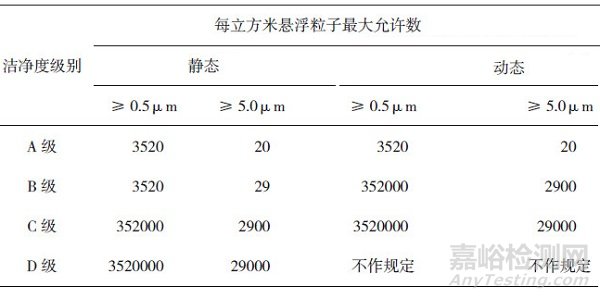
表 3 不同等级洁净环境中悬浮粒子最大允许数
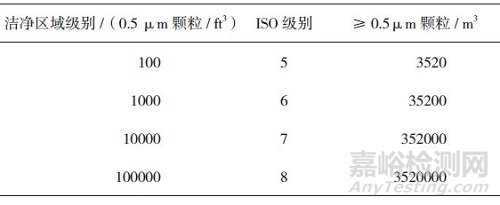
表 4 不同等级洁净环境中悬浮粒子最大允许数(美国)
我国GMP和药包材标准、欧盟GMP、PIC/S和日本对于浮游菌、沉降菌和表面接触菌检测标准规定一致, 动态标准规定见表 5。美国GMP中对于浮游菌和沉降菌标准规定见表 6, 美国GMP中没有对于表面接触菌的规定。

表 5 不同等级洁净环境微生物允许数

表 6 不同等级洁净环境微生物允许数(美国)
各标准和法规对于其他参数标准规定的具体内容见表 7。PIC/S组织主要由欧洲国家及澳大利亚、加拿大等国家组成, 欧盟GMP的附录通常来自于PIC/S推荐的GMP指导文件, 澳大利亚也使用PIC/S文件用于指导药品生产[26]。由于美国不是PIC/S的成员国, FDA起草的GMP指导文件通常不会参考PIC/S的GMP指导文件, 因此, 在各项标准中, 欧盟与PIC/S的GMP许多内容一致, 但美国则有自己的标准体系。
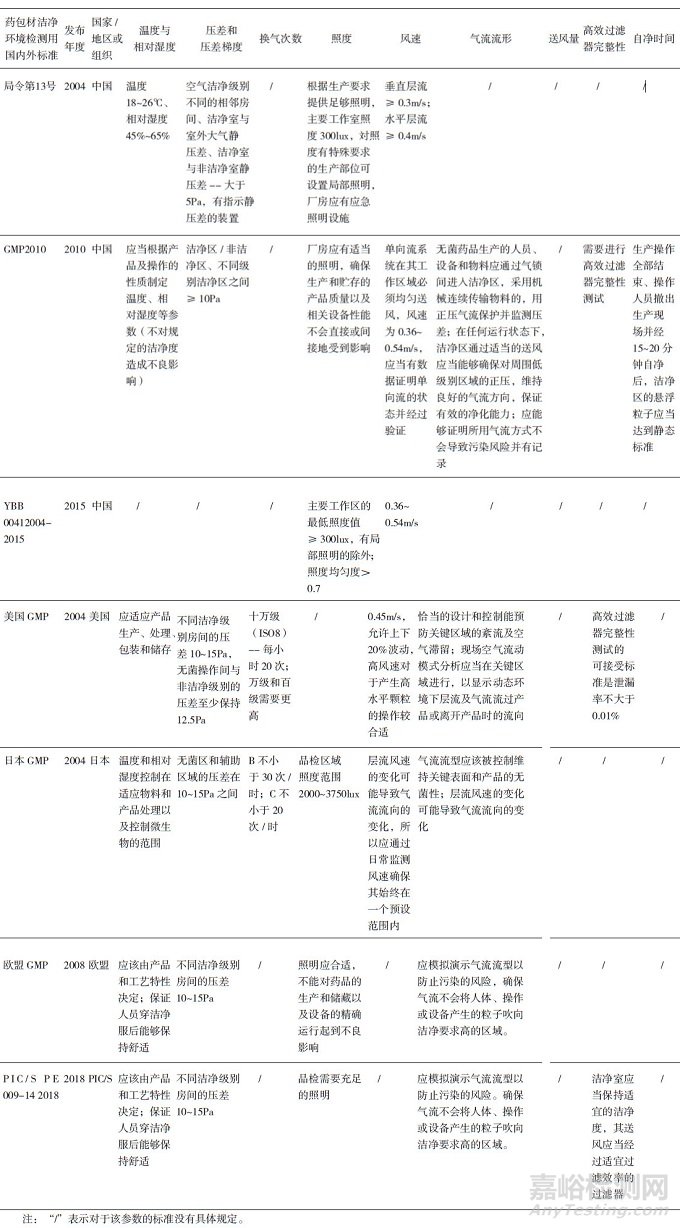
表 7 国内外洁净环境法规和标准对于各参数标准的规定
4 质量控制的异同
培养基模拟灌装试验是无菌药品生产管理的重要组成内容[27], 在培养基模拟灌装测试的规定方面, 我国GMP与欧盟GMP的规定基本一致, 要求发生任何微生物污染均应进行调查。美国和PIC/S对于培养基模拟灌装测试也均有表述。而我国的局令第13号和药包材标准在无菌工艺中的培养基模拟灌装方面还是空白。
在洁净环境的验证方面, 局令第13号、我国GMP和药包材标准、PIC/S、美国以及澳洲的GMP中均有相关表述。但区别是, 国外相关标准和法规已经把验证和确认作为GMP的核心内容, 而我国法规和标准的相关表述则仅是为了确认厂房和设施设备正常运行。
另外, 质量风险管理是GMP的精髓, 对于质量风险管理的规定, 我国和其他各国的GMP关于质量风险管理的表述内容一致, 均参照人用药物注册技术要求国际协调会(ICH)指南[28]。而我国药包材标准是把风险管理结合到了测试项目中进行表述, 局令第13号中并未对质量风险管理进行描述。
5 讨论与建议
国外法规将药品包装材料和药用辅料与所对应的药品生产纳入同一个管理和要求体系, 洁净生产厂房的相关要求一致。我国2010年修订的GMP已与欧盟GMP看齐, 我国药包材标准中的药包材洁净环境测试方法与我国GMP总体保持一致, 在高效过滤器完整性和气流流型等的测试上, 方法标准已经建立, 但是缺少判断标准, 在自净时间和表面接触菌等的测试方法和标准上仍是空白, 也缺乏培养基模拟灌装试验的规定。局令第13号由于形成时间较早, 缺乏一些参数细节上的规定。
国外的相关法规和标准在不断更新, 欧盟已在2017年12月发布GMP附录一"无菌药品的生产"新草案, 目前仍在广泛征求意见阶段, 修改的内容包括强调开发基于风险评估的系统, 明确洁净环境等级确认与日常监测的区别, 细化粒子与微生物的环境和生产监测等, 相较于我国目前的GMP, 内容更加精细和完善。我国技术管理部门应关注国际上药包材洁净环境检测方面的动态, 结合我国药包材行业的发展特点, 对相关法规和标准进行及时的补充和更新。
目前, 我国药包材与药品关联审评审批管理的模式, 也对药包材生产和使用企业的洁净环境提出了更高的要求。企业的洁净室在设计施工时, 即应严格参照GMP的规定, 根据产品特征设计生产车间的洁净环境, 使其建筑装饰、风系统、气体系统、水系统、物料供应系统等符合规范, 生产过程中按照要求使用, 并进行洁净环境的维护[8, 29]。另一方面, 在实际使用中定期监测才能保证洁净室达到设计指标和使用要求, 但目前许多生产企业在洁净环境中开展日常监控和参数验证工作的频次和程度不够, 在培养基灌装等一些微生物控制上仍存在一定程度的缺陷, 并没有将标准严格执行到位[6, 30]。根据药包材洁净环境的相关法规和标准规定, 建议药用包材的生产企业根据洁净度级别和空气净化系统确认的结果及风险评估情况, 建立有规律且符合频次要求的动态监控方案, 加强洁净区的日常监控和参数验证的工作。另外, 要对各级别洁净环境的各关键参数设定预警限值, 对于异常情况及时调查和纠偏, 排查原因, 增强洁净环境的综合控制水平[31]。
现阶段我国洁净环境检测行业的整体水平参差不齐, 存在对检测标准的执行和实践力度不够的现象。由于技术水平的限制, 在检测过程中也会出现"假阴性"结果, 即无法检测出洁净环境真实存在的问题。该问题提示洁净环境检测行业应加强对相关法规和标准的执行度, 并不断强化检测技术和水平, 推进行业的发展和进步。另一方面, 我国相关监管部门也应加强对药包材生产环境的监管力度, 采取多种形式的抽查和检查, 督促企业提高生产环境洁净级别, 加强其日常监控和验证工作, 尽快做到在与药物制剂相匹配的洁净环境中生产, 最大程度降低或避免对药物制剂质量的影响。
药包材是药品的重要组成部分, 药包材的质量问题会直接增加药品的安全风险, 我国药包材生产的洁净环境, 应在多部门和行业的协作下, 尽快实现真正的高标准、严要求, 成为药品整体质量的一个重要保障。
参考文献
[1] 国家食品药品监督管理局.国家食品药品监督管理局令第13号直接接触药品的包装材料和容器管理办法[S]. 2004.
[2] 中国药典: 四部[S]. 2015: 通则413(药包材通用要求指导原则).
[3] 刘恕. 关联审评审批制度下药品生产企业如何做好药包材的质量控制[J]. 中国医药工业杂志, 2018, 49(8): 1191-1194.
[4] 中国食品药品检定研究院. 国家药包材标准[M]. 北京: 中国医药科技出版社, 2015: 312-323.
[5] 杨会英, 赵霞, 贺瑞玲, 等. 药包材关联审评审批申报资料技术要求的对比分析[J]. 中国药事, 2018, 32(3): 310-316.
[6] 孙世昌. 信息化背景下浅谈药厂洁净室污染控制措施[J]. 科学与信息化, 2018(17): 172, 175.
[7] 国家食品药品监督管理总局. 2016年第134号公告关于药包材药用辅料与药品关联审评审批有关事项的公告[S]. 2016.
[8] 中华人民共和国住房和城乡建设部, 国家质量监督检验检疫总局. GB 50591-2010洁净室施工及验收规范[S]. 2010.
[9] 国家质量监督检验检疫总局, 国家标准化管理委员会. GB/T 16292-2010医药工业洁净室(区)悬浮粒子的测试方法[S]. 2010.
[10] 国家质量监督检验检疫总局, 国家标准化管理委员会. GB/T 16293-2010医药工业洁净室(区)浮游菌的测试方法[S]. 2010.
[11] 国家质量监督检验检疫总局, 国家标准化管理委员会. GB/T 16294-2010医药工业洁净室(区)沉降菌的测试方法[S]. 2010.
[12] 国家食品药品监督管理局.药品生产质量管理规范[S]. 2010.
[13] 国家质量监督检验检疫总局, 国家标准化管理委员会. GB/T 36066-2018洁净室及相关受控环境检测技术分析与应用[S]. 2018.
[14] U.S. Food and Drug Administration. Sterile Drug Products Produced by Aseptic Processing-Current Good Manufacturing Practice Chapter 4: Buildings and Facilities[S]. 2004. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/sterile-drug-products-produced-aseptic-processing-current-good-manufacturing-practice
[15] Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme. PIC/S PE 009-14 2018 Guide to Good Manufacturing Practice for Medicinal Products[S]. 2018.
[16] Pharmaceutical and Medical Device Agency, Japan. Japanese Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing-Chapter 7: Processing Areas for Sterile Pharmaceutical Products[S]. 2004.
[17] Pharmaceutical and Medical Device Agency, Japan. Japanese Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing-Chapter 6: Buildings and Facilities[S]. 2004.
[18] European Commission Enterprise and Industry DirectorateGeneral. EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use - Chapter 3: Premises and Equipment[S]. 2014.
[19] European Commission Enterprise and Industry DirectorateGeneral. EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use - Annex 1: Manufacture of Sterile Medicinal Products[S]. 2008.
[20] Department of Health Therapeutic Goods Administration, Australian Government. Australian Code of Good Manufacturing Practice for Medicinal Products[S]. 2012.
[21] International Standardization Organization. ISO14644- 3: 2005 Cleanrooms and Associated Controlled Environments-Part 3: Test Methods[S]. 2005.
[22] International Standardization Organization. ISO14644-2: 2015 Cleanrooms and Associated Controlled Environments -Part 2: Monitoring to Provide Evidence of Cleanroom Performance Related to Air Cleanliness by Particle Concentration[S]. 2015.
[23] International Standardization Organization. ISO14644- 1: 2015 Cleanrooms and Associated Controlled Environments-Part 1: Classification of Air Cleanliness by Particle Concentration[S]. 2015.
[24] International Standardization Organization. ISO 14698-2: 2003 Cleanrooms and Associated Controlled Environments-Biocontamination Control-Part 2: Evaluation and Interpretation of Biocontamination Data[S]. 2003.
[25] International Standardization Organization. ISO 14698-1: 2003 Cleanrooms and Associated Controlled Environments-Biocontamination Control-Part 1: General Principles and Methods[S]. 2003.
[26] 张卓光. PIC/S、PIC/S GMP与中国GMP的比较研究[J]. 中国药事, 2010, 24(3): 302-305.
[27] 吴美杰, 王连群, 张春峰, 等. 培养基模拟灌装试验的结果观察与样品管理[J]. 机电信息, 2018(29): 46-48. DOI:10.3969/j.issn.1671-0797.2018.29.010
[28] 郝赟, 余哲, 王唤雨, 等. 欧盟GMP附录15确认与验证解读[J]. 机电信息, 2017(11): 1-4, 17. DOI:10.3969/j.issn.1671-0797.2017.11.001
[29] 张玉萍. 洁净厂房环境中的给排水体系设计[J]. 四川水泥, 2014(8): 129. DOI:10.3969/j.issn.1007-6344.2014.08.111
[30] 毛红. 培养基模拟灌装试验相关缺陷分析与改进建议[J]. 中国药业, 2018, 27(18): 80-82. DOI:10.3969/j.issn.1006-4931.2018.18.026
[31] 袁桂淦. 制药企业洁净室洁净度影响因素分析及控制[J]. 养生保健指南, 2017(48): 173. DOI:10.3969/j.issn.1006-6845.2017.48.167

来源:xml-data


