您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-03-07 17:19
一、基本概况
1.自然环境
哈萨克斯坦位于亚洲中部。北邻俄罗斯,南与乌兹别克斯坦、土库曼斯坦、吉尔吉斯斯坦接壤,西濒里海,东接中国。国土面积为272.49万平方公里。多数居民信奉伊斯兰教(逊尼派)。此外,还有东正教、天主教和佛教等。1991年12月16日哈萨克斯坦共和国宣布独立。
2.人口和行政区划
哈萨克斯坦人口为2016万(截至2024年8月)。约有140个民族,其中哈萨克族占70.6%,俄罗斯族占15.1%。哈萨克语为国语,官方语言为哈萨克语和俄语。
全国设17个州、3个直辖市(阿斯塔纳、阿拉木图、奇姆肯特)。首都为阿斯塔纳(Astana)。
3.2024年出口概况
2024年,中国向哈萨克斯坦出口医疗器械总计30.25亿元人民币,同比增长18.66%。
二、监管机构和法规要求
国家药品医疗器械专业技术中心 (National Center for Expertise of Medicines and Medical Devices)负责哈萨克斯坦的医疗器械产品注册。哈萨克斯坦医疗器械注册需要遵循第KR-DSM-10号令《药品和医疗器械审查规则的批准》法规要求。
三、医疗器械定义
◆ 医疗器械:用于提供医疗服务的材料、产品、溶液、试剂、试剂盒、套装等。
◆ 医疗设备:按照制造商设定的功能用途和性能特性,用于单独或组合提供医疗服务的装置、仪器、设备、组件或系统。
◆ 体外诊断试剂:体外诊断医疗产品是指用于医疗目的的任何仪器、装置、设备、材料、试剂、校准物、控制材料及其他产品,这些产品可以单独使用,也可以彼此组合使用,还可以与必要的附件共同使用以实现预期用途,包括专用软件。这些产品由医疗产品制造商设计用于对人类生物材料样本进行体外研究,以获取以下信息:生理或病理状态、先天性疾病、对某种临床状况或疾病的易感性、与潜在接受者的组织相容性、对治疗效果的反应预测、治疗药物的选择及(或)治疗监测。
四、产品分类
1.分类依据
◆ 分类依据:
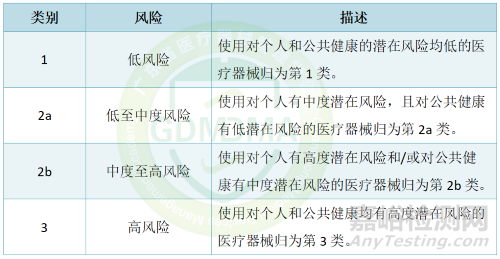
根据No. ҚР ДСМ-281/2020号法令,医疗器械及IVD的风险等级分类如下。
2.分类
3.分类标准
◆ 医疗器械:
1)使用时长:医疗器械的使用时间长短。
2)侵入性:医疗器械与人体接触或进入体内的程度。
3)与人体接触:医疗器械是否与人体接触或与人体相连。
4)引入方式:医疗器械通过人体自然腔道或手术方式进入体内。
5)对重要器官和系统的影响:医疗器械是否作用于心脏、中央循环系统或中枢神经系统等关键系统。
6)能源使用:医疗器械运行中涉及的能源类型及程度。
◆ 体外诊断试剂:
1)用于检测血液、血液成分、血液衍生物、细胞、组织或器官中感染因子的体外诊断医疗器械,以评估其输血或移植的可能性,以及用于识别导致高传播风险的威胁生命疾病感染因子的医疗器械,归为第3类。
2)用于确定血型或组织类型以保证血液、血液成分、细胞、组织或器官输血或移植免疫相容性的体外诊断医疗器械,归为第2b类。但AB0系统(A (AB01)、B (AB02)、AB (AB03))、Rh系统(Rh1 (D)、Rh2 (C)、Rh3 (E)、Rh4 (c)、Rh5 (e))、Kell系统(Kell (K))、Kidd系统(JK1 (Jka)、JK2 (Jkb))和Duffy系统(FY1 (Fya)、FY2 (Fyb))归为第3类。
3)以下用途的体外诊断医疗器械归为第2b类:识别性传播疾病感染因子;在脑脊液或血液中检测中度传播风险的感染因子;检测感染因子,若错误结果可能导致患者或胎儿死亡或残疾;对孕妇进行筛查以确定其对感染的免疫状态;确定感染病状态或免疫状态,若错误结果可能导致危及生命的治疗决策;癌症的选择性治疗、分期、筛查或诊断;遗传测试;监控药物、物质或生物成分水平,若错误结果可能导致危及生命的治疗决策;治疗患有威胁生命的感染病患者;筛查胎儿先天性疾病。
4)用于自测的体外诊断医疗器械归为第2b类。若通过此类器械获得的测试结果不涉及关键医学状态或为初步结果且需与相应实验室测试对比,则归为第2a类。
5)没有测量功能、客观特性为一般实验室使用的体外诊断医疗器械,且被制造商明确用于体外诊断过程(不指定具体类型的实验室测试/分析物),以及用于生物样本容器的医疗器械,归为第1类。
6)不适用以上规则的体外诊断医疗器械,包括具有测量功能且执行未固定实验室测试列表(依赖所用试剂盒)的分析设备,归为第2a类。设备与所用试剂的相互依赖性不影响其分类为第2a类。
五、注册流程
1.注册流程
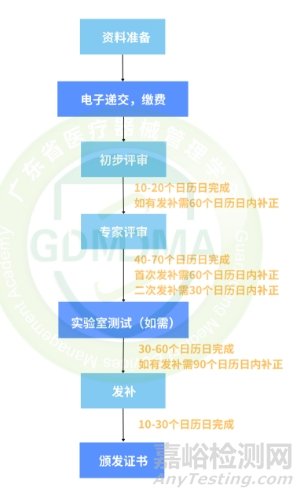
1)初步审评:评估申请人在注册资料中提交的文件的完整性、齐全性以及真实性是否符合现行法律要求。如果注册资料中存在任何问题,申请人将收到一封电子数字签名认证的信函,信函中列明发现的问题以及需在不超过60个日历日内全部解决的要求,否则会收到医疗器械审查终止的通知。
2)实验室测试:
由国家专家机构的检测实验室进行,通过物理、化学、生物和技术测试进行,目的是确认医疗器械是否符合制造商质量文件中声明的安全性和质量指标(生物安全性或生物作用评估、物理和机械指标、功能、技术以及物理化学指标),包括以下内容:
A.分析技术和法规文件中有关检测方法的部分;
B.验证是否符合质量文件的要求;
C.确定分析质量控制方法的可重复性。
3)专家评审:注册资料中确认医疗器械安全性、质量和有效性的文件进行专家评估和分析;
针对1类和2a类医疗器械的审查:专业审查:40个日历日(包括确认包装布局、标签、贴纸、使用说明的标识真实性或翻译成哈萨克语);
针对2b类(高风险)和3类(最高风险)医疗器械的审查:专业审查:70个日历日(包括确认标识真实性或翻译成哈萨克语)。
4)在以下情况下不进行实验室检测:
A.医疗设备;
B.提供来自公告机构的文件,证明医疗器械生产和产品质量控制体系完全符合欧洲委员会关于医疗器械的指令要求;
C.对在欧盟和/或英国、美国、加拿大、日本或瑞士的制造商生产场所制造的医疗器械进行审查,且该医疗器械已获得欧盟、英国、美国、加拿大、日本或瑞士监管机构批准流通;
D.医疗器械的重新注册;
E.医疗器械的加速审查。
5)医疗器械的加速审查:总时间不超过30个工作日;
以下情况适用:产品再次注册、军事行动及其后果的消除、突发事件的发生、预防及消除后果、特别危险传染病的发生、传播威胁以及其后果的消除。
2.注册提交技术文件

3.注册周期及费用
注册周期约6-9个月,费用400USD-1000USD/张注册证。具体费用可进入官网计算。
4.注册提交流程
注册提交通过集成的国家专家组织的信息系统进行,申请人可以通过账户获得注册状态信息。向国家专家机构的申请服务中心(以下简称“CSC”)提交以下文件:
1)以电子和纸质形式提交医疗器械注册申请;
2)包含医疗器械注册资料清单的注册资料包,以电子形式提交;
3)申请人支付审查费用至国家专家机构银行账户的付款凭证;
4)供实验室检测用的医疗器械样品,提供化学品标准品、微生物测试菌株、细胞培养物、特定试剂以及实验室检测方法所需的消耗品,这些物品数量应足以完成三次测试,且剩余有效期不少于六个月,并符合存储和运输条件,除非制造商的质量文件另有规定。
5.现场审核要求及注意事项
2a无菌类、2b类及3类需要接受现场审核。
6.该区域有关UDI的要求
无特殊要求。
其他注意事项或特别提醒
1)需原产国注册证。
2)哈萨克斯坦注册证永久有效,2025年仍可以按哈萨克斯坦本土法规提交注册,注册费用及周期远低于EAEU注册的费用。

来源:广东省医疗器械管理学会


