笔者最近在整理我国近年来间充质干细胞产品的IND注册申报情况时发现,越来越多的企业开始布局干细胞治疗这一赛道。国家药品监督管理局药品审评中心(NMPA)仅2023年受理的干细胞产品就有42项,获批数目就达到24项,还有14个产品尚在评审阶段。间充质干细胞产品呈井喷式爆发,与其经众多临床数据验证所展示出的强有力的治疗效果和产品的安全性密不可分。而干细胞产品从工艺探索、药学研究再到临床转化的阶段,整体生产流程中需要遵循严格的质量控制。本文将干细胞产品药学研究全流程分成主要的三个阶段,按照生产用原材料的采集(组织/细胞采集),多级细胞库的制备以及终产品(原液/半成品/制剂)的生产,分阶段介绍关键的质量研究内容,包括过程中涉及的质量评价项目、关键质控点和参考质量标准及实施案例。
一、组织/细胞采集阶段(以脐带源间充质干细胞为例):
作为生产用起始原材料,也是整个产品开发的开端,需要严格进行供者管理。在脐带采集前,需签订供供者捐献知情同意书用于伦理审查;在采集脐带时,需要筛选供者年龄、无传染性疾病、无遗传性疾病,在供者健康调查表中需统计过去家族病史与现病史。在采集脐带的同时,可以同时采集产妇静脉血与脐带血用于病毒检测作为物料入场控制。
在脐带的采集、运输、接受整个过程中需保证无菌;人源病毒检测项主要包括:HIV、HAV、HBV、HCV、HTLV、EBV、CMV、HPV、B19、人疱疹病毒6/7/8、人多瘤病毒、腺病毒、SARS-CoV-2等。
本阶段检测方法多参考药典法条,无菌检测通常采用药典法中的需/厌氧菌培养法及薄膜过滤法。染色体核型检测方法为G带染色。人源病毒检测方法通常为凝集法、ELISA或Q-PCR法。
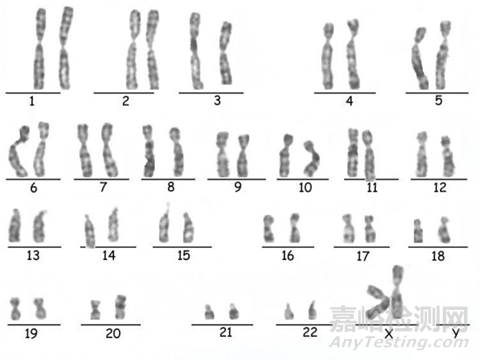
图1:G带染色法-染色体核型(正常为46,XX或46,XY)
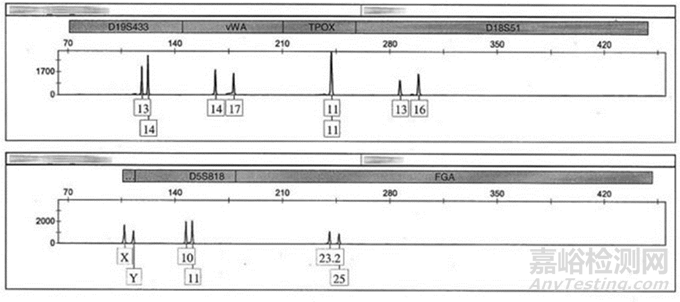
图2:STR图谱分析报告
本阶段质量要求:1、无菌检查呈阴性2、产妇血病毒为阴性3、染色体核型正常4、STR无细胞间交叉污染。
二、关键中间体(多级细胞库)的制备阶段
不同的工艺路线,其关键中间体也有所不同。多数组织来源的间充质干细胞通常会建立主细胞库与工作细胞库。在细胞冻存前会进行放行检验,只有符合放行质量标准的样品才可以用于建库保存。此时检测项目包括细胞的生物学活性(细胞活率和细胞数、细胞增殖能力检测)、鉴别(细胞形态观察、细胞表面标志物)以及微生物安全性(无菌检测、支原体检测、外源病毒检测、端粒酶活性与染色体核型检测等)。
在细胞冻存入库后,为了保证冻存的细胞能够满足后续的生产要求,还要抽库进行更全面的质量检测。主要检项包括:生物学活性(细胞活率和细胞数、细胞增殖能力)、细胞鉴别(细胞形态观察、细胞表面标志物)、生物学安全性检查(体外成瘤性、STR、染色体核型、端粒酶活性)、生物学效能(淋巴细胞增殖抑制能力、针对特定适应症体外药效调节)、微生物安全性检查(无菌、支原体)等。
与此同时,考虑到维持各级细胞库在长期冻存过程中的质量属性,需深入研究细胞库的冻存稳定性,考察现有冻存条件对细胞质量的影响,尤其是长期冻存稳定性(时间点包括3个月、6个月、一年甚至更久)。稳定性研究期限至少需涵盖上市前临床试验期限的要求,考察项目与细胞库全检检项相同。
本阶段检测方法与结果分析举例:
1、细胞形态观察
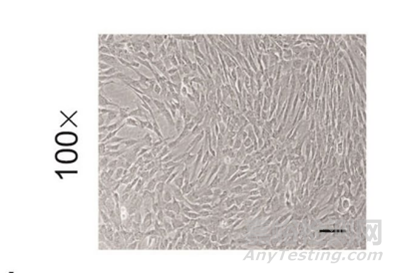
图3:显微镜细胞形态观察:应为贴壁、长梭形、旋涡状生长[1]
2、细胞表面标志物检测
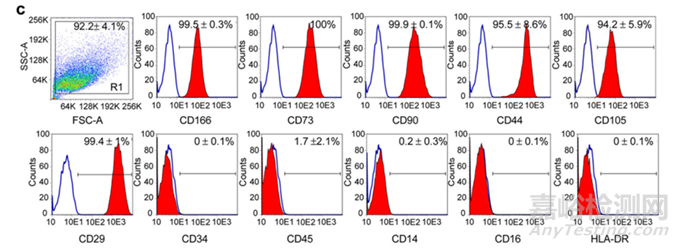
图4:“三阳五阴”流式表型检测[2]:需符合可接受标准,可扩展标志物如CD166/CD44/CD133等
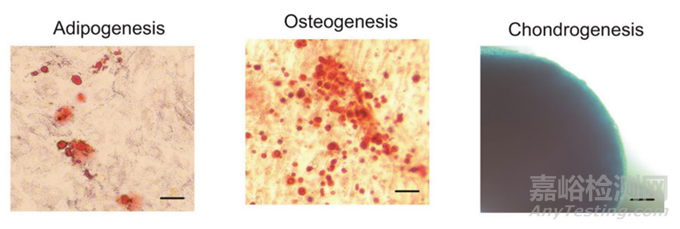
3、三系分化能力考察
图5:成脂、成骨、成软骨球分化照片[1]
对于三系分化能力的检测通常可做染色法、Q-PCR法以及WB法鉴定诱导后特异性标志物的表达。推荐提供Q-PCR检测特征性基因(成骨:ALP、Runx;成脂:LPL、PPARγ;软骨球:Collagen II等)代替WB法。
4、细胞数量、活率与细胞群体倍增时间检测
细胞数量与活率常使用细胞计数仪,经台盼蓝或AO/PI双荧光染色,统计细胞活率并读取细胞数,推算细胞收获量。
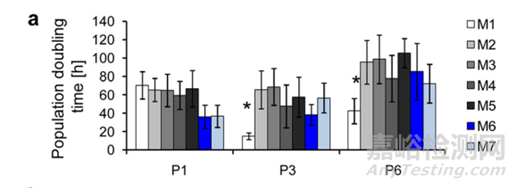
关于细胞的增殖能力,主要通过绘制生长曲线推算细胞群体倍增时间(PDT,Population Doubling Time)。PDT计算公式= [log2/(lgNt-lgN0)] ×T,公式中T为细胞达到平台期的培养时间(96 h), Nt为t时间收获细胞数,N0为起始接种细胞数。
图6:细胞PDT统计[2]
5、端粒酶活性检测
在遗传学安全性方面,对细胞端粒酶活性的检测也很关键。大多数肿瘤等恶性增殖细胞具有端粒酶活性,正常的体细胞中端粒酶常常失活。hTERT亚基的mRNA水平基因表达量可用来衡量端粒酶活性。Q-PCR检测结果应为阴性,样品即合格。
6、MSC免疫调节能力
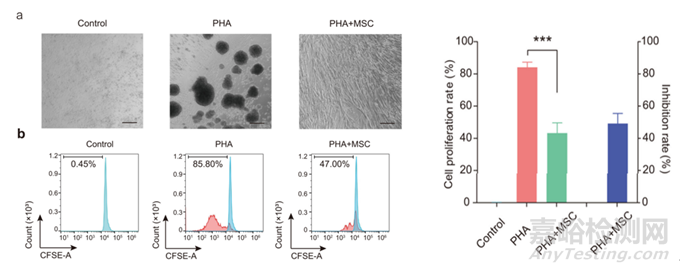
图7:MSC抑制人淋巴细胞增殖功能[1]
MSC具有抑制人淋巴细胞增殖的能力,通过CFSE标记人PBMC活细胞,随细胞分裂荧光强度逐步减半,通过流式检测CFSE荧光强度推算子代与亲代细胞比例,能够检测细胞的增殖能力。通常将不同浓度的MSC与预处理激活的PBMC细胞共培养多天,计算PBMC细胞增殖抑制比例。这里需要优化共培养试验条件后,测试多批不同来源的PBMC,计算合适的抑制率放行范围。这里不同来源的PBMC引起的结果波动比较大,可以对使用的PBMC批号做提前筛选,以免影响判定结果。
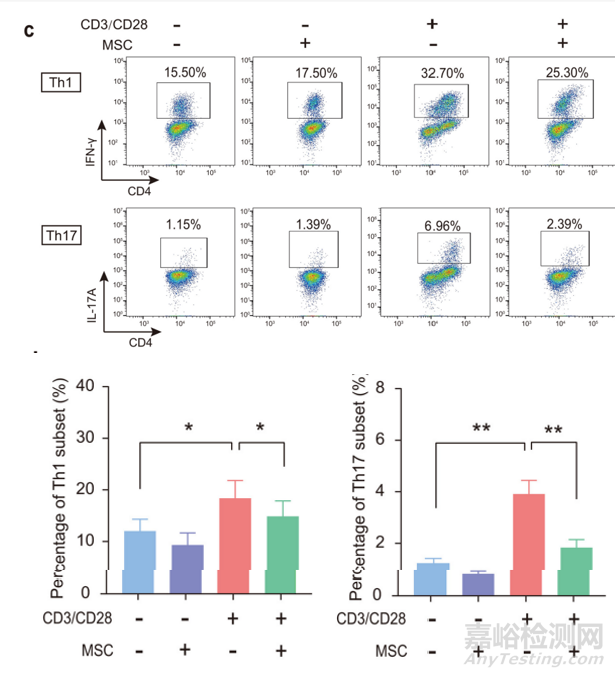
图8:MSC调节人淋巴细胞亚群增殖功能[1]
除抑制淋巴细胞增殖外,MSC还能够抑制淋巴细胞Th1和Th17亚群。
图9:MSC调节人淋巴细胞分泌细胞因子的能力[1]
也可以选择图中提供的多种细胞因子,如发挥抑炎作用的IL-10,促炎因子TNFα、INFγ等,检测MSC对免疫细胞分泌的细胞因子含量的影响,评估产品的免疫调节能力。
对细胞库的质量标准(可参考):
1、生物学活性:细胞活率不低于80%,增殖能力细胞倍增时间在20-30 h;
2、细胞鉴别:形态观察为长梭形、成纤维细胞样,细胞表面标志物流式结果应满足ISCT最低检定标准的CD73/90/105+>95%,CD19/34/45/11b、HLA-DR+<2%,具有向成骨、成软骨、成脂分化能力;
3、生物学安全性检查:体外成瘤实验应无成瘤,STR检测无细胞间交叉污染,染色体核型正常,端粒酶活性应为阴性;
4、微生物安全性检查:无菌/支原体检查应为阴性,人源病毒检测应为阴性;
5、生物学效能考察:间充质干细胞对淋巴细胞增殖抑制功能应为抑制,对淋巴细胞分泌细胞因子功能应为抑制。
三、对原液和制剂的质量控制
对于终产品的质量也需要做全面检测。在辅料加入前,对原液的质量检测包括:理化性质(渗透压检测)、细胞鉴别(形态与活率数量、染色体核型、STR、流式表面标志物表型)、生物学效能(三系分化能力、细胞因子分泌、免疫抑制能力等)、生物学安全性(软琼脂克隆形成、端粒酶活性)、外源成分残留(冻存液DMSO、BSA、庆大霉素等)和微生物检测(无菌、支原体、内毒素、人源病毒检测)。
原液通过加入辅料以及灌装后形成制剂。相应的检项在此时也最为全面,包括理化特性(渗透压、pH、产品外观、可见异物及装量)、细胞鉴别(形态与活率数量、染色体核型、STR、流式表面标志物表型、同工酶、STR、细胞周期、凋亡及衰老、细胞倍增时间及克隆形成率等)、生物学效能(三系分化、细胞因子分泌、免疫细胞抑制调节和不同疾病细胞模型的药效调节)、生物学安全性(软琼脂克隆形成、端粒酶活性)、残留检测(DMSO、TrypLE消化酶、残余培养基)、微生物安全性(支原体、无菌、厌氧/需氧菌、内毒素、人源性病毒检测)。作为输注入人体前的最后一道关卡,对制剂的检项最为全面,能够保证在临床应用时的安全性。
这里需要注意,产品与工艺相关杂质可结合稳定合理的工艺清除验证进行分析控制。不同工艺过程中,对终产品中存在残留并且可能影响干细胞产品质量和安全有效性的工艺相关杂质,例如BSA、消化酶、冻存液成分,或者有些工艺中涉及分选和三维条件下微载体培养等,应关注相关成分残留检测做为放行控制。
对制剂的质量标准(可参考):
1、理化:产品外观应符合标准,pH范围在6.0-8.0,渗透压范围在1500~2000 mOsmol/kg,可见异物不超限,装量应符合产品规格;
2、杂质:死细胞比例应低于25%,杂细胞比例<2%,DMSO含量应≤8 ug/mL,消化液含量应≤15ng/mL ,培养基残留量应≤0.125mg/mL;
理化和杂质为制剂阶段增加的检项,此外对细胞鉴别、安全性检查、生物学效能的检测项与质量标准与前文对细胞库的质量标准相同,可参考前文内容。
干细胞生产过程中不可避免的需要经过传代,可能会存在基因组特性的转变,随着传代代次增加,其遗传不稳定性风险也越高,也可能造成细胞异质性。根据《中国药典》2020年版对于生物制品稳定性试验的指导原则,还需要对包括传代稳定性、制剂加速与长期保存稳定性、制剂模拟运输条件稳定性、联合使用稳定性等进行考察以保证产品质量可控。传代稳定性需要明确体外生产限制传代的代次与临床上使用的细胞代次,在确定的工艺代次基础上继续传代并进行细胞生长鉴别与遗传稳定性考察。制剂的加速和长期保存研究则需要对制剂在不同取样时间(4h、8h、12h、24h等)和温度环境(4℃、25℃、37℃避光等条件)考察相关指标。运输稳定性研究中,考虑到模拟运输条件、冷链保存和使用过程中的常规与极端情况等,可以假设运输过程为颠簸路面,运输时间在8-12 h间,且在开始运输和运输终点的首尾脱离冷链暴露于25℃环境。还需要进行制剂联合使用稳定性的研究,在制剂制备后到临床使用期间,按照冷链保存-运输-临床使用等场景,考察整个过程对细胞的质量是否存在影响。药企还需制定产品上市批准后的稳定性研究计划,用来保证药品批准上市后的主细胞库、工作细胞库、原液与制剂的长期稳定性。
四、小结
本文按照干细胞产品开发流程,煎蛋梳理了过程中的关键质量研究内容,并对过程中涉及的质控点、评价项和可参考的质量标准做了举例说明。细胞治疗产品因为全程使用的活的细胞,因此对其安全性、生物学有效性的考量更加严格。在产品开发过程中,深入学习药典和CDE发布的指导原则将更好的帮助我们开发出稳定、有效且安全的产品。
五、参考资料
1、Stem Cell Reviews and Reports DOI/10.1007/s12015-023-10556-8
2、J Mol Med (2017) 95:205–220 DOI 10.1007/s00109-016-1471-7
3、细胞治疗产品研究与评价技术指导原则(试行)(2017年12月22日发布)
4、人源干细胞产品药学研究与评价技术指导原则(试行)(2023年4月25日发布)
5、中国药典2020版三部、四部-生物制品稳定性试验的指导原则
6、免疫细胞治疗产品药学研究与评价技术指导原则(试行)(2022年5月26日发布)