摘 要 / Abstract
本文利用ClinicalTrials.gov数据库信息,简要分析4家大型医疗器械外资企业(美敦力、强生、雅培及波士顿科学)近年来在我国开展上市前临床试验的现状,并结合相关政策法规的进展,探讨政策法规对外资企业在我国开展临床试验的影响以及展望未来的发展趋势。结果显示,在筛选的52个临床试验研究中,最早的研究从2011年开始,2011~2022年4家外资企业在我国开展上市前临床试验的数量总体呈现增长趋势,但近3年有所下降。52个临床试验研究涉及的产品以心血管领域为主,单臂研究居多,样本量中位数为82,大部分研究时限不超过12个月。从分析结果可以看出,政策法规对外资企业在我国开展临床试验具有决定性的影响。
Using data from the ClinicalTrials.gov website, this article briefly analyzes the pre-market clinical trials conducted in China during recent years by four major foreign medical device companies(Medtronic, Johnson & Johnson, Abbott and Boston Scientific). Considering the progress of relevant regulations and policies, it explores the impact of relevant policies on the participation of foreign companies in pre-market clinical trials in China and offers insights into potential future trends. The analysis of 52 selected clinical trials reveals a trend of increasing numbers from 2011 to 2022 for these four companies. However, there has been a decrease in the past three years. The 52 selected clinical trials predominantly focus on cardiovascular products, often adopting single-arm study designs with a median sample size of 82 and a follow-up period of fewer than 12 months. The findings underline the significant impact of policies and regulations on clinical trials conducted by foreign medical device companies in China.
关 键 词 / Key words
医疗器械;外资企业;临床试验;注册;上市前
medical device; foreign company, clinical trial; registration; pre-market
过去20多年,我国医疗器械监管实践发生了较大的变化,对医疗器械临床试验的要求也在不断发展之中。2000年,《医疗器械监督管理条例》[1]发布实施,并于2014年和2021年经历了两次重大修订[2-3],与其配套的相关规定也随之进行了多次修订与完善[4-6]。此外,还发布了一系列涉及具体产品的指导原则,其中与外资企业密切相关的指导原则,包括《接受医疗器械境外临床试验数据技术指导原则》[7]等。
临床试验是评价产品安全性和有效性的重要途径,对于高风险产品更是不可或缺的环节。但由于临床试验耗时耗力耗资,企业在进行临床试验前需反复权衡,慎重决策,特别是在境外已有临床数据证实产品安全性和有效性的基础之上。基于此,本文简要分析了近年来美敦力、强生、雅培及波士顿科学4家大型医疗器械外资企业在我国开展上市前临床试验的现状,并结合相关政策法规的进展,探讨相关政策对外资企业在我国开展临床试验的影响以及展望未来的发展趋势。
1、方 法
本文数据主要来自美国临床试验注册库(Clinical Trials.gov)[8]。该数据库是由美国食品药品监督管理局(Food and Drug Administration,FDA)和美国国立医学图书馆(National Library of Medicine,NLM)共同开发,并于2002年正式对公众开放[9]。尽管该数据库设定了一些格式化的条目及定义,但具体查找和搜索时仍可能存在不同的理解,且有些信息不够完整甚至可能存在差错,进而影响相关研究分析的准确性。因此,本文明确了筛选上市前注册临床试验的判定标准:(1)明确表明是上市前临床试验研究的可直接入选;(2)对于未明确表明是上市前临床试验研究的,通读整个记录并结合特定产品在我国上市的情况以及其他行业内的信息进行综合判断。此外,本文还通过检索中国临床试验注册中心相关数据,核实是否存在遗漏信息,以保证研究的准确性。
在Clinical Trials.gov数据库的高级检索“Sponsor(Lead)”栏中分别输入美敦力、强生、雅培及波士顿科学4家医疗器械外资企业的英文名称(Medtronic、Johnson&Johnson、Abbott、Boston Scientific),考虑到强生还有较多子公司,又将其子公司的英文名称(Biosense Webster、Cordis、De Puy、Ethicon、Life Scan、Vision Care)进行输入;在“Country”栏中选择China(中国)或在“Other terms”栏中输入China(中国);检索日期为2000年1月1日~2022年12月31日。按照筛选标准逐条阅读分析检索记录,剔除误检记录(如检索Johnson&Johnson时会出现Mead Johnson)后共获得206个临床试验研究。再剔除药物、体外诊断试剂、上市后研究以及研究中心包含中国香港而不包含中国内地的记录等,最终有52个临床试验研究符合本文分析要求,其中包括6个在海南博鳌开展的真实世界研究项目。
2、结 果
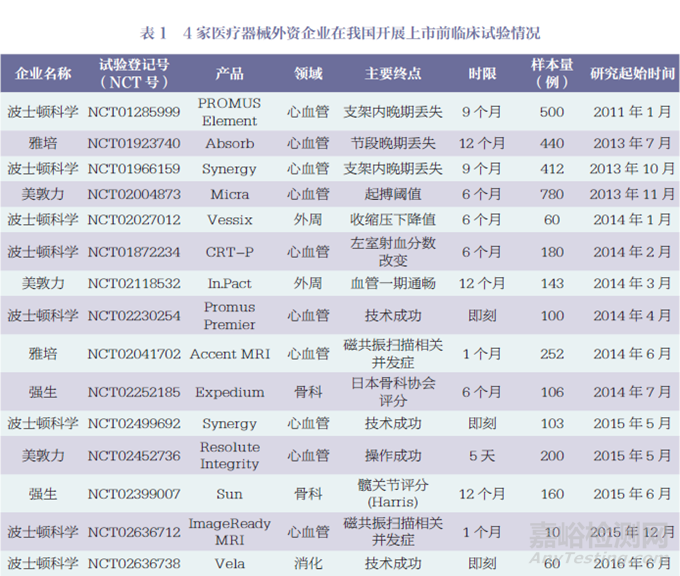
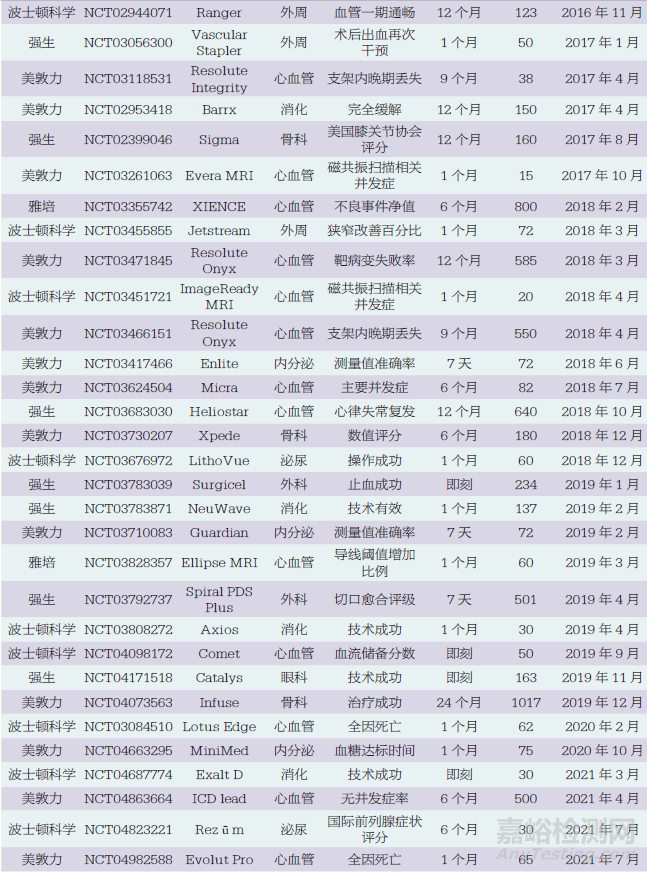
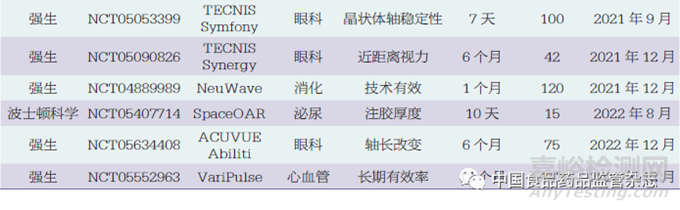
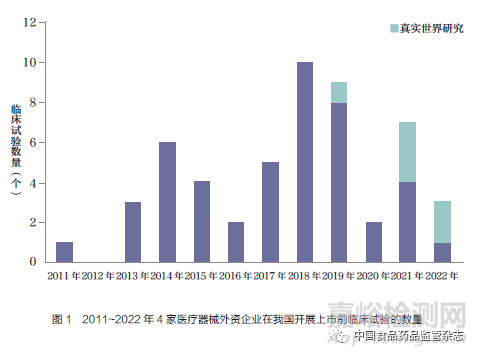
本文对4家医疗器械外资企业在我国开展上市前临床试验情况进行统计,结果显示,在52个临床试验研究中,其中波士顿科学18个、美敦力16个、强生14个、雅培4个,如表1所示。最早的研究从2011年开始,2011~2022年4家医疗器械外资企业在我国开展临床试验的数量总体呈现增长趋势,但近3年有所下降,如图1所示。
按医疗器械治疗领域分类,52个临床试验研究中,心血管内科24个(冠状动脉11个、心脏起搏9个、电生理2个、心脏瓣膜2个),消化介入6个,外周血管领域5个,骨科5个,眼科4个,内分泌3个,泌尿外科3个,外科2个。冠状动脉领域中10个为冠状动脉支架,心脏起搏领域中5个为磁共振兼容起搏器或植入型心律转复除颤器(implantable cardioverter defibrillator,ICD)。
按研究设计分类,52个临床试验研究中,单臂研究38个,随机对照试验(randomized controlled trial,RCT)14个,其中包括一致性评价研究1个。
52个临床试验研究中,主要终点的随访时间范围为即刻至24个月。具体为:即刻7个,1个月及以下20个,1个月以上至12个月以下15个(主要为6个月和9个月),12个月9个,12个月以上1个。
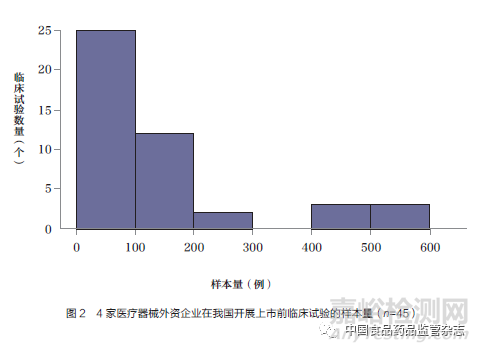
52个临床试验研究中,国际多中心临床研究7个,包括2个亚太地区多中心临床研究。2个亚太地区多中心临床研究中,我国内地临床研究中心数量/亚太地区多中心临床研究中心总数分别为1/13和3/21;其他5个国际多中心临床研究中,我国内地临床研究中心数量/国际多中心临床研究中心总数分别为1/56、2/51、2/41、3/27、2/46。除了这7个研究外,其他45个研究在我国内地开展的试验样本量最小为10例,最大为585例,中位数为82(四分位间距60~160),如图2所示。其中,样本量为200例以上的有8个,包括6个冠状动脉支架产品和2个外科产品。
本文纳入了6个在海南博鳌开展的真实世界研究,即Catalys飞秒激光(强生)、Exalt D一次性十二指肠镜(波士顿科学),Rezūm前列腺水蒸气热疗消融系统(波士顿科学),Space OAR水凝胶(波士顿科学),ACUVUE Abiliti人工晶体(强生),TECNIS Synergy人工晶体(强生)。尽管这6个研究为真实世界研究,但考虑到其支持产品注册的性质,故纳入分析范围。除此之外,52个临床试验研究中有5个研究提前终止,其中2个因入选困难,3个因企业战略变化。
综上所述,共有52个临床试验研究纳入本文分析,其中波士顿科学18个、美敦力16个、强生14个、雅培4个。研究起始时间从2011年开始,2011~2022年4家医疗器械外资企业在我国开展上市前临床试验的数量总体呈现增长趋势,但近3年有所下降。临床试验涉及的产品以心血管领域为主,单臂研究居多,样本量中位数为82,大部分临床研究时限不超过12个月。
3、讨 论
2000年发布的《医疗器械监督管理条例》尚未明确提出临床试验的表述,相关表述为临床试用和临床验证。2004年发布的《医疗器械注册管理办法》和《医疗器械临床试验规定》[10]开始使用临床试验的表述,2014年修订的《医疗器械监督管理条例》删除临床试用和临床验证的表述。2016年发布的《医疗器械临床试验质量管理规范》[11]第三条将临床试验定义为:在经资质认定的医疗器械临床试验机构中,对拟申请注册的医疗器械在正常使用条件下的安全性和有效性进行确认或者验证的过程。2022年修订的《医疗器械临床试验质量管理规范》[12]删除临床试验定义中的“或者验证”。
2004年发布的《医疗器械注册管理办法》在附件12中规定:境外政府医疗器械主管部门未批准在本国(地区)上市的第二类、第三类产品,以及境外政府医疗器械主管部门已批准申请产品在本国(地区)上市但企业无产品进入过我国市场的第三类植入型产品,需要在我国境内进行临床试验。因此,在较长的一段时间内未检索到外资企业在我国开展上市前临床试验的注册记录,直至2011年才出现冠状动脉药物支架的上市前临床试验。
2014年修订的《医疗器械监督管理条例》及《医疗器械注册管理办法》明确提出:申请第二类、第三类医疗器械注册,应当进行临床试验(另有免于进行临床试验的医疗器械除外)。2018年发布的《冠状动脉药物洗脱支架临床试验指导原则》[13]指出:境外已上市产品如已经在境外完成设计良好的、前瞻性的临床试验,同一试验方案下试验组样本量不少于600例且符合我国对于该类产品的临床试验要求,除应按相关规定提供产品境外的临床资料外,还应提供产品在中国境内开展的随机对照试验研究资料。并建议冠状动脉药物洗脱支架确证性试验由两个临床试验组成,其中一个临床试验为随机对照试验,另一个临床试验为单组目标值试验。其中随机对照试验为与对照产品进行的以晚期管腔丢失(Late Loss)为主要研究终点的1:1的不少于200对的试验;单组目标值试验以靶病变失败率(TLF)为主要研究终点,样本量应不少于800例,其中部分病例可来源于随机对照试验的试验组。本文列选的临床试验研究中有5个冠状动脉支架开展了200对以上的试验,实际上还有上市后临床试验的要求但是不在本文讨论范围内。
对于心脏瓣膜类产品,2019年发布的《经导管植入式人工主动脉瓣膜临床试验指导原则》[14]要求境外已上市产品在我国开展临床试验,但样本量要求较低,也未强调随机对照试验。具体要求为:在有境外良好数据的前提下,在我国境内需开展样本量不少于50例的临床试验。本文列选的临床试验研究中有2个主动脉瓣,其样本量分别为62例和65例。
2018年发布的《接受医疗器械境外临床试验数据技术指导原则》更规范、科学,旨在为申请人通过医疗器械境外临床试验数据申报注册以及监管部门对该类临床试验数据的审评提供技术指导,避免或减少重复性临床试验,加快医疗器械在我国上市进程。从国家药品监管部门公布的相关数据来看,已经有一些产品通过该途径获批在国内上市,免于国内临床试验。此外,为了使相关单位更好地理解关于如何界定的不同因素对临床数据产生有临床意义影响,该指导原则特意列举了2个具有人群差异的产品,即脉搏血氧仪设备和用于遗传病基因检测的体外诊断试剂。
2020年发布的《真实世界数据用于医疗器械临床评价技术指导原则(试行)》[15]在可考虑将真实世界证据用于医疗器械临床评价的常见情形中列出:临床急需进口医疗器械在国内特许使用中产生的真实世界数据,可用于支持产品注册,作为已有证据的补充。当前在海南博鳌开展的临床真实世界数据应用试点工作正是该项政策的重要体现,因此本文也纳入了截至2022年12月的6个医疗器械真实世界研究项目。
4、展 望
本文纳入的临床试验以心血管领域的产品为主,特别是冠状动脉药物支架,因药物支架是以医疗器械为主的药械组合产品,因产品的特殊性,国家药品监管部门要求相关单位在我国境内开展临床试验。从临床实践的角度来看,目前全球范围内冠状动脉药物支架的使用尚未发现具有明显的人群差异,未来随着临床实践大量数据的积累,境外已上市产品在充分的境外临床数据基础之上,是否可以减少或者免除临床试验,值得研究人员关注与探讨。
2018年发布的《接受医疗器械境外临床试验数据技术指导原则》,要求在提供境外数据时应分析技术审评要求的差异、受试人群的差异及临床试验条件的差异,其中较难把握的是受试人群的差异。国家药品监督管理局医疗器械技术审评中心网站的审评论坛中有两个问答可以为上述难点提供一些思路:一个是“通过临床试验路径开展临床评价时,指导原则中要求除提供境外临床试验数据,仍需在中国境内开展临床试验,申请人是否需开展临床试验”,回答为“《接受医疗器械境外临床试验数据技术指导原则》第四条中已明确‘若特定医疗器械的技术审评指导原则中含有对其临床试验的相关要求,该器械境外临床试验应考虑有关要求,存在不一致时,应提供充分、合理的理由和依据’。因此,若申请人已经按照《接受医疗器械境外临床试验数据技术指导原则》提交了符合伦理、依法、科学原则的临床试验数据,且充分考虑了技术审评要求的差异、受试人群的差异、临床试验条件差异,可不在中国境内额外开展临床试验。”另一个是“《需进行临床试验审批的第三类医疗器械目录》中的产品是否可以用境外临床试验资料进行申报?临床试验是否还需在中国境内进行审批”,回答为“根据2018年1月发布的《接受医疗器械境外临床试验数据技术指导原则》,列入《需进行临床试验审批的第三类医疗器械目录》(下称《目录》)的医疗器械,亦可按照上述指导原则要求用境外临床试验数据进行申报。对于产品境外临床试验资料不符合相应要求,仍需在中国境内进行临床试验的《目录》中产品,临床试验仍需审批后方能开展。”这两个解读可为相关企业提供临床试验工作思路,减轻企业负担,节约社会资源,同时也能使患者及时获益。相关文件的修订与发布规范了我国临床试验的发展方向,这也符合本文发现近年来外资企业在我国开展上市前临床试验数量下降的趋势。
目前,我国参与国际多中心临床研究上市前临床试验的案例并不多,其中开展临床试验的前期准备时间较长是重要原因之一。此外,如果产品检验及人类遗传资源审批这两个节点能有所突破(如2021年修订的《医疗器械监督管理条例》对自检报告的认可),我国同步参与全球研究的项目将会有所增长。
是否开展临床试验,对于企业来说是重大的决策。2021年发布的《决策是否开展医疗器械临床试验技术指导原则》《医疗器械临床评价技术指导原则》[16]等系列文件,对于相关企业具有重要的指导意义。但是在实际操作层面上,可能还是会存在各方理解的偏差。因此,希望在是否需要开展临床试验及对临床试验等关键的问题上监管部门能开展更多的沟通,以减少临床试验中的不确定性。
医疗器械更新换代速度快,这对监管部门和企业来说都是挑战。是否需要开展临床试验(特别是在有境外临床数据的基础上)及如何开展临床试验,将是监管部门、企业及研究人员持续关注的问题。国家药品监管部门一直在探索前进,外资企业在国内开展临床试验的图景将逐渐明晰起来,相信未来将会有更多的安全有效的医疗器械引入我国,造福患者。
引用本文
曾治宇,韩磊,张晓星,彭琳,曾理.关于医疗器械外资企业在我国开展上市前临床试验的探讨[J].中国食品药品监管,2023(8):38-45.