重庆市药品监督管理局关于征求《关于优化医疗器械说明书变更管理要求的通知(征求意见稿)》修改意见的通知,内容如下:
重庆市药品监督管理局关于优化第二类医疗器械说明书变更管理要求的通知(征求意见稿)
各检查局、市药审中心、市药品监测中心、重庆医疗器械质量检验中心、审批处、器械监管处、各医疗器械注册人:
为进一步落实“放管服”改革和优化营商环境要求,推动医疗器械产业高质量发展,优化医疗器械说明书变更管理要求,按照《医疗器械监督管理条例》、《医疗器械注册与备案管理办法》、《医疗器械说明书和标签管理规定》等法律法规规定,现将第二类医疗器械(不含体外诊断试剂)说明书变更管理有关问题通知如下:
一、根据风险评估进行分类管理
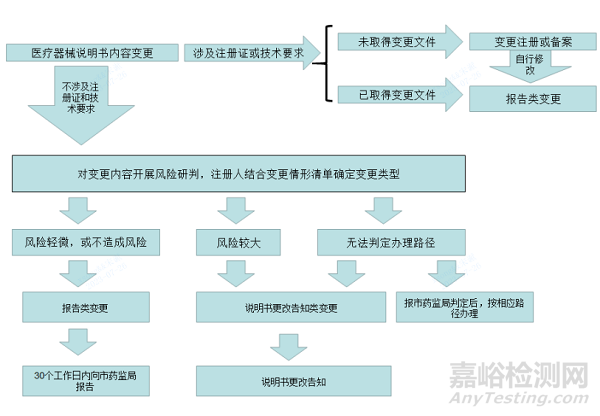
不涉及到产品注册证和产品技术要求内容的医疗器械说明书变更分为说明书更改告知类变更和报告类变更。说明书变更经风险研判可能带来一定产品质量风险或使用风险的属于说明书更告知类变更,应办理医疗器械说明书更改告知。说明书变更经风险研判不造成新的风险或仅带来轻微的产品质量风险、使用风险的属于报告类变更。注册人对报告类变更应在质量管理体系控制下保存设计变更记录,可不提出说明书更改告知申请,但应按照《医疗器械注册与备案管理办法》第七十九条规定,在30个工作日内向药品监督管理部门报告。
二、按照报告类变更管理的主要变更情形
(一)根据《医疗器械说明书和标签管理规定》第十六条相关规定,已注册的医疗器械办理了变更注册或变更备案的,申请人应当在取得变更文件后,依据变更文件自行修改说明书。
(二)借鉴参照《总局办公厅关于体外诊断试剂说明书文字性变更有关问题的通知》(食药监办械管〔2016〕117号)有关规定,以下情形按照报告类变更管理:
1. 医疗器械注册人或者生产企业联系方式、售后服务单位名称及联系方式、生产许可证编号的变化,注册人应在相关信息变化后,自行修改。生产许可证编号应在相应省级药监部门发放生产许可证后再行修改。
2. 医疗器械说明书“医疗器械注册证编号/产品技术要求编号”项目,在药监部门发放医疗器械注册证后,导致该项内容变化的情况,注册人应自行修改。
3. 医疗器械说明书中列明必须配套使用的医疗器械,由于监管部门发放医疗器械注册证/备案凭证后,导致说明书中载明的配套使用的医疗器械或体外诊断试剂注册证编号/备案凭证编号发生变化的情况,注册人应自行修改。
4. 说明书中所用的图形、符号、缩写等内容的解释变化,因注册人按照YY/T 0466.1-2016《医疗器械 用于医疗器械标签、标记和提供信息的符号 第1部分:通用要求》、YY/T 0466.2-2015《医疗器械 用于医疗器械标签、标记和提供信息的符号 第2部分:符号的制订、选择和确认》等标准完善相应标识的解释内容,导致该项内容变化,但不涉及其他需办理注册变更的情况,注册人应自行修改。但根据《关于GB 9706.1-2020及配套并列标准、专用标准实施有关工作的通告》解读之二,医用电气设备类产品应在完成新版GB 9706系列标准变更注册后,修改产品说明书和标签。
(三)说明书的版式、颜色、尺寸、企业标识、注册商标、防伪标识条码、医疗器械唯一标识、企业网址等非医疗器械注册批准证明性内容。
(四)医疗器械说明书的修订日期等不涉及与医疗器械相关的文字内容变化的。
(五)发现医疗器械说明书个别文字编排、书写错误或遗漏,项目有颠倒或遗漏的。
(六)说明书中以下内容变更,注册人在医疗器械质量管理体系控制下按照设计开发变更的管理要求,经风险评估,不增加产品质量风险和使用风险的:
1. 安装和使用说明变更,且变更后的表述更加细化和完善的。
2. 产品维护和保养方法与原表述相比更加严格细致的,特殊储存、运输条件、方法与原要求相比更加严格细化的。
3. 新增禁忌症、注意事项、警示以及提示的内容。
三、按照说明书更改告知类变更管理的常见变更情形
(一)产品技术要求中未载明的物理、化学等性能参数或技术内容的变更。
(二)减少或变更禁忌症、注意事项、警示以及提示的内容。
(三)产品维护和保养方法变更,且与原表述相比更加简化的,特殊储存、运输条件、方法与原要求相比更加宽松的。
(四)配件清单的变更,包括配件、附属品、损耗品更换周期以及更换方法的说明。
(五)重复使用医疗器械在重复使用时的处理过程变更,如清洁、消毒、包装及灭菌的方法和重复使用的次数或者其他限制。
(六)产品外形、外观的变更。
(七)产品图示的变更,包括标识、接口、操控面板、应用部分等细节。
(八)产品包装说明、储存运输稳定性的说明、使用期限的变更。
(九)产品交付状态(是否无菌)说明的变更。
(十)预期使用环境、适用人群的变更。
(十一)与本产品联合使用实现预期用途的其他医疗器械或非医疗器械产品的详细信息的变更。
(十二)治疗类产品的量效关系和能量安全性说明的变更。
(十三)软件类医疗器械或医疗器械的软件组件未体现在产品技术要求中的相关内容变更。如:软件标识、必要的显示窗口,人工智能医疗器械的算法基本信息、数据收集、算法训练、算法性能评估,与其他医疗器械或非医疗器械交换并使用信息的基本信息、需求规范、风险管理、验证与确认、维护计划,采用移动计算、云计算、虚拟现实等信息通信技术实现预期功能与用途的基本信息、需求规范、风险管理、验证与确认、维护计划等内容。
(十四)产品技术要求中未载明但体现在说明书中的产品原材料信息的变更。
(十五)与患者直接或间接接触的器械,未在产品技术要求中体现但体现在说明书中与生物学评价相关的信息。
(十六)安装和使用说明变更,且变更后的表述更加简化的,或变更后与变更前相比安装使用说明变化显著可能带来新的使用风险的。
四、工作要求
(一)注册人落实产品质量主体责任,对变更内容开展风险研判并参考前述变更情形确定变更类型后,按照相应路径办理(办理路径示意图见附件)。注册人无法准确判定的,应按照说明书更改告知类变更办理或报市药监局判定。
(二)完善医疗器械说明书变更审核的技术支撑机制。医疗器械说明书更改告知事项办理过程中,经研判可能带来较大风险的,根据实际情况由市药审中心或重庆医疗器械质量检验中心提出技术意见作为重要办理参考。市药审中心加强医疗器械说明书源头审核管理,不断提升医疗器械说明书内容规范性。
(三)本文件在执行过程中遇到的问题应及时反馈,以便于及时开展会商评估和优化完善。本文件的规定与国家药监局后期出台政策不一致的,以国家药监局相关规定为准。
附件:
医疗器械说明书内容变更办理路径示意图