摘 要 Abstract
为了给药品上市后监管提供技术支撑,按照国家药监局统一部署,国家药监局药品审评中心制定了《已上市生物制品药学变更研究技术指导原则(试行)》,并于2021 年6 月发布,受到了广泛关注。本文详细论述了在基于风险进行分类管理的上市后监管制度下,药品质量体系在变更中的重要作用,明确了基于变更进行分类管理及开展相关研究的基本原则,并对几个具有代表性的变更事项的分级及需要开展的技术研究进行了说明,以期加强指导原则的指导作用。
In order to provide technical support for regulation of post-approval pharmaceutical changes of biological products, the Center for Drug Evaluation drafted the Technical Guideline for Post-approval Pharmaceutical Changes of Biological Products (Trial), which has received extensive attention since its official release in June 2021. This paper discusses in detail the important role of pharmaceutical quality system in change management under post-marketing regulatory system and the basic principles of classification management and related research based on the change classification. In addition, this paper explains the classification of several representative change items and the technical research needed to be carried out, so as to strengthen the guiding role of the guiding principle.
近年来, 随着《药品管理法》《疫苗管理法》《药品生产监督管理办法》《药品注册管理办法》《药品上市后变更管理办法(试行)》等一系列法律法规的颁布实施,我国药品监管领域法规体系建设更加科学和现代化,对于药品的管理更加强调全生命周期管理,对于已上市生物制品的药学变更,逐渐由既往基于事项的变更转变为基于风险进行分类管理的模式,并且强调了药品上市许可持有人(以下简称持有人)是上市后变更管理的责任主体。这种监管模式的转变更有利于激发企业上市后自主变更的主动性,使企业可以更灵活地基于风险和企业实际进行上市后药学变更管理,从而有助于实现生物制品的全生命周期管理,保障生物制品上市后供应。为了给药品上市后监管提供技术支撑,按照国家药监局统一部署,国家药监局药品审评中心起草了《已上市生物制品药学变更研究技术指导原则(试行)》(以下简称《变更指导原则》),明确了药学变更分类及开展相关技术研究等内容[1]。笔者在此对在《变更指导原则》起草、宣贯过程中公众普遍关心或者比较疑惑的上市后变更问题进行论述,以供参考。
1、稳健的药品质量体系是基于风险的上市后变更管理的前提
药品质量体系(pharmaceutical quality system,PQS)的建立和完善贯穿于药品的整个生命周期。在全生命周期的各个阶段, 使用基于科学和风险的方法对生物制品进行管理,可促进整个生命周期的持续改进。稳健的PQS 不仅能保证在受控的药品生产质量管理规范(Good Manufacturing Practice of Medical Products,GMP) 条件下获得符合要求的产品,还意味着持有人具备较为完善的知识管理和质量风险管理能力。具备有效的PQS,是实施生物制品上市后药学变更的前提和必要条件[2]。对于生物制品的上市后药学变更,PQS 的重要作用具体表现在以下方面。
首先,持有人通过对生物制品前期研发资料、变更前开展的工艺验证进行研究,以及对既往生产经验和通过变更管理所获得的产品和工艺知识的理解,有助于更好地判断拟开展变更的风险。
其次,持有人应科学识别、评估和控制潜在的变更风险,从而合理地开展基于风险的变更分级和变更研究。
再次,上市后变更分为审批类变更、备案类变更和报告类变更,所有上市后药学变更验证都应当符合GMP 要求, 在PQS的变更管理体系中完成。
最后,持有人在变更完成后获得的变更知识有助于促进工艺性能和产品质量持续改进,从而进一步强化PQS。
需要注意的是,生物制品上市后药学变更情形复杂、种类繁多,无法只通过一个技术指导原则来涵盖所有变更情形。实际中存在大量变更需要在已经建立的PQS 内将持有人作为上市后变更的责任主体,根据内部变更分类原则、工作程序和风险管理标准,并结合产品特点,参考有关技术指导原则,在充分研究、评估和必要验证的基础上确定变更管理类别。
2、上市后药学变更分类及考量
基于风险的上市后变更管理已在部分国家和地区实施多年,鉴于我国尚缺乏足够的经验,在《变更指导原则》起草过程中,起草小组广泛查阅了国际人用药品注册技术协调会(the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)、世界卫生组织(World Health Organization,WHO)、美国食品药品监督管理局(Food and Drug Administration,FDA)、欧洲药品管理局(European Medicines Agency,EMA)等组织或机构发布的基于风险进行变更分类的相关指南,并结合我国既往变更监管实际,对常见的生物制品上市后药学变更事项进行了分类列举。这一方面体现了现阶段我国对变更事项的风险认知,另一方面也符合我国的监管实际。
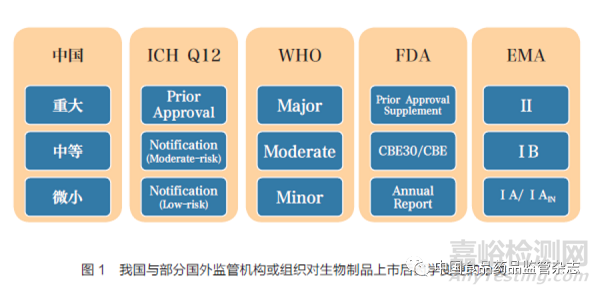
《变更指导原则》按照对安全、有效和质量可控的风险和产生影响的程度,将已上市生物制品药学变更依次分为重大变更、中等变更和微小变更。我国与部分国外监管机构或组织对生物制品上市后变更的分类如图1 所示[3-7]。根据《药品注册管理办法》,变更应当相应地以补充申请、备案和年度报告的方式进行管理。除重大变更、中等变更和微小变更外,通过以上方式进行管理的还有其他变更事项,如对于新增制剂规格等需要发放新药品批准文号的上市后变更,隶属国家药监局职责范围,应当按照《药品注册管理办法》相关要求申报补充申请[8]。此外,部分药学变更事项对生物制品的物质基础产生影响的可能性较大,如疫苗佐剂变更,应考虑按照《生物制品注册分类及申报资料要求》中改良型相关类别进行申报[9]。为了充分体现基于风险的管理理念,随着生物制品行业的发展以及持有人内部管理经验的积累,持有人在确保内部PQS 稳健的前提下,可以根据《药品上市后变更管理办法(试行)》对目前《变更指导原则》中已经确定的分类级别进行降级管理,同时应与省级药品监管部门沟通,并在达成一致意见后实施。对于疫苗、细胞治疗产品等生物制品的上市后变更,除参考《变更指导原则》开展研究外, 另有规定和技术要求的, 也应遵照执行。
3、上市后药学变更研究
《药品上市后变更管理办法(试行)》规定,药品上市后变更不得对药品的安全性、有效性和质量可控性产生不良影响。对于任何生物制品的上市后药学变更,均需要开展相应的变更研究来支持变更。
一般情况下,持有人可以通过线性缩小规模对拟变更的工艺参数、新物料等开展研究,初步确定拟变更的参数范围,但变更可比性研究一般应在商业化规模下完成。生物制品变更可比性研究的开展可参考ICH Q5E[10]。可比性研究是基于风险、数据驱动的全面评价,开展变更可比性研究是进行已上市生物制品药学变更评价的基础和关键。对于已上市生物制品的药学变更,产品可比性的确定可以药学研究为基础。但当现有药学数据对于确定可比性而言不充分时,应开展相关的非临床和(或)临床研究来获得补充性证据,可能涉及的情况包括:可比性研究结果显示变更前后存在差异,但持有人无充分的数据来证实该差异对于安全性、有效性无不良影响;药学变化影响较大或部分生物制品本身较复杂时,仅通过药学研究无法全面评价变更对安全性、有效性的影响;变更前的药学研究数据缺失,无法确定可比性验收标准,无法评价药学可比性。在开展桥接研究方面,原则上,如果非临床研究数据足以弥补药学研究数据的不足,从而能够获得可比性结论时,则无需开展临床研究。但对于部分生物制品,由于缺乏合适的动物模型,或动物模型与人体数据相关性不确定时,可能需要开展适宜的临床桥接研究来确定变更的影响。
在开展变更可比性研究时,研究批次与变更风险程度相关。通常情况下,针对重大变更,至少需要利用变更后的工艺开展连续3 批商业化规模验证。在该阶段,持有人一方面应根据工艺参数执行情况和中间体监测数据等,对变更后的工艺稳健性、外源因子控制能力、生产物料的循环次数、中间体的暂存时间等进行确证,另一方面应利用通过工艺验证获得的产品开展质量和稳定性可比性分析,从而既能确保变更后工艺控制能力不降低,又能证明变更前后的产品具有可比性。在验证批次数量方面,某些情况下可考虑适当减少验证批次。例如,当存在多种不同装量规格时,将同样的原液分别灌装不同装量的制剂,若原液工艺完全一致,持有人可考虑在原液阶段开展至少连续3 批验证,在制剂阶段结合装量差异,可以考虑采用括号法、矩阵法等来减少验证批次。同时,建议持有人提前与监管机构进行沟通交流,通过提供充分的依据支持括号法或矩阵法的使用。
在目前我国实施的基于风险进行变更分类的变更管理模式下,持有人作为变更责任主体,其变更管理能力的高低直接决定变更管理的成败。对于已经具有较完备的PQS 的企业,现有的变更管理模式可以使持有人更加自主、灵活、高效地结合实际开展变更管理;但对于变更风险识别经验不足、既往药品研发及生产中积累的工艺和产品相关知识缺乏的持有人,若不能全面评估和识别变更风险,将会导致变更分类错误,进而导致变更研究不足,从而造成变更风险不可控。因此,若持有人无法全面评估变更风险,建议参照《变更指导原则》,按照较高级别的要求开展相关研究,获得的研究结果一方面有助于持有人对变更进行评估,另一方面积累了企业内部知识,有利于对产品全生命周期的变更管理。
4、生物制品上市后药学变更分类示例及技术要求解读
4.1 表达载体的变更
利用重组表达技术生产的生物制品(包括重组蛋白制品、重组蛋白制品衍生物)通常需要在原核或真核表达系统中导入稳定的表达载体,选择理想的单克隆后,建立三级种子库或细胞库,然后利用工作种子批或细胞库进行发酵生产。因此,一旦变更表达载体,通常需要重新建立细胞库,并需要对后续一系列工艺参数(可能包括生产用原材料)进行必要的调整,影响范围广,且导致产品翻译后修饰等质量属性发生改变的可能性较大。由于该类变更风险较大,凡是涉及表达载体变更的均属于重大变更。但即便是重大变更,表达载体的变更也有前提条件,即目的蛋白和宿主细胞均未改变。设置该前提条件的含义包含两个层面:一方面,改变表达载体的同时不能改变宿主细胞,是因为二者同时变更的影响程度和风险会进一步升高,不仅会对目的蛋白本身的质量属性造成影响,还会对残留宿主蛋白、残留宿主DNA 等杂质的种类和残留量造成影响,仅通过变更研究可能无法全面评估其对产品安全性、有效性的影响,需要重新申报新的临床试验来评估产品的安全性、有效性;另一方面,表达载体变更时不能对目的蛋白的序列造成改变,即不能增加或删除标签、不能改变蛋白质氨基酸序列,但通过同义突变对目的基因序列进行密码子优化以提高表达量,不影响目的基因最终翻译成的氨基酸序列是被允许的。
持有人需要按照《变更指导原则》中相关技术要求开展变更研究和可比性分析。需要说明的是,《变更指导原则》中“表达载体、种子批及细胞库”变更类别对应的1~14 条技术要求,并非对所有品种均适用。例如,第11条技术要求规定,“如涉及,更新种子批/ 细胞库质量标准。提供变更种子批/ 细胞库质量标准的依据和检验结果。”若持有人未变更内部的种子批/ 细胞库的验收标准,则无需提供相关研究数据。只有发生了关联变更的,持有人才需要提交相关数据。如果表达载体变更后,同时伴随了生产工艺的变更,持有人应当对相关变更进行明确,提供变更前后工艺流程及工艺参数的对比内容,并且应当按照关联变更的要求提供工艺变更的支持性数据。应特别注意的是,一旦工艺参数变更影响了病毒去除/ 灭活工艺,持有人还应开展病毒去除/ 灭活工艺再验证,并重新参考ICH Q5A[11]的要求分析产品的病毒安全限度。同理,如果伴随设备变更、生产用物料变更等,要参照《变更指导原则》相关变更事项,针对各个变更分别开展变更研究,同时商业化规模的可比性研究可以一并开展。
根据质量源于设计(quality by design,QbD)的研发理念,原则上,在产品开发阶段,持有人应当从全生命周期的角度,选择最适宜的表达载体。考虑到表达载体变更风险较大,不鼓励在上市后特别是在持有人对工艺和产品知识积累不足时开展表达载体变更。但随着监管政策完善、科技进步、行业发展等,某些产品确实面临着必须变更表达载体的问题。例如,针对去除β- 内酰胺类抗生素抗性,持有人应当开展充分、全面的可比性研究,并且,考虑到表达载体变更的风险较高、影响范围较广,通常建议持有人开展适当的非临床/ 临床桥接研究。
4.2 生产场地的变更
生产场地的变更可能涉及管理法规问题,持有人需要根据相关法律法规要求,确保产地变更符合现有的相关法律法规要求,参考《变更指导原则》进行变更研究、确定变更级别。对于疫苗的生产场地变更,应同时参考《疫苗生产场地变更质量可比性研究技术指导原则》[12] 相关要求。
由于生物制品场地变更可能伴随设备、物料、人员的变更,可能会对生产工艺及控制、质量控制体系、产品质量等产生影响,通常认为变更风险较大。在《变更指导原则》中,涉及原液生产场地的变更包括重大变更和中等变更两个级别;涉及制剂生产场地的变更包括重大变更、中等变更和微小变更三个级别。其中,原液和制剂生产场地的中等变更均需要满足一定的前提条件。设置相关前提条件的考虑因素主要包括:①变更前后的生产线(含生产用设备、生产用原材料等)属于完全复制,这样的变更不会引起工艺参数的变化、不会因为生产用原材料的改变而引入额外的外源因子风险或新杂质残留、不会因为直接接触材料的改变而引入新的浸出风险、不会导致外源因子风险升高(如收获液中逆转录颗粒数量不受影响)或无菌控制风险升高;②变更前后的生产场地应受控于同一质量体系,无论是自主生产还是委托生产,持有人均应承担产品质量控制体系主体责任,应确保变更前后生产场地在工艺性能和产品质量监测、纠正和预防措施、变更管理和管理回顾等方面均按照持有人建立的质量体系规定执行;③对于不同类型生物制品(如血液制品、传统疫苗、治疗用重组蛋白类产品),无论是在GMP 要求、生产工艺方面,还是在产品本身质量控制方面,均差别较大,若已有产自新生产场地的同类产品获批,那么在GMP 执行、生产工艺控制及产品质量监测方面就有可借鉴的经验,因此相对风险较低。若不能满足上述条件,原液或制剂生产厂/ 厂房/ 生产线的变更均应按照重大变更管理。另外,虽然在满足前提条件的情况下,在已获批的制剂生产场地增加包装线/ 贴签线属于微小变更,但若涉及批准文件相关地址的修改,应按照备案管理。
在《变更指导原则》中,对于不同的变更类别,生产场地变更研究的技术要求存在差异。以原液生产场地变更为例, 重大变更涉及的技术研究内容包括1~18、21 技术要求,而中等变更涉及的技术研究内容包括1~6、8、10、13、16、17 技术要求。由于中度变更不涉及规模、工艺、生产用物料等的变更,因此无需重新对细胞传代稳定性进行研究,无需开展病毒去除/ 灭活工艺再验证,无需重新开展色谱填料循环次数的研究,无需重新对直接接触材料的相容性进行研究,无需开展不同设备变更所需的相关研究。若无特殊规定,除非现有药学研究数据对于确定可比性研究而言不充分,一般情况下,中等变更的技术研究内容不涉及非临床/ 临床桥接研究。
4.3 生产工艺的变更
在《变更指导原则》中,工艺变更主要包括三大模块:生产规模、工艺程序和步骤、工艺参数。所有工艺变更都应当证明变更后工艺控制能力不低于变更前工艺控制能力,否则就不符合《药品上市后变更管理办法(试行)》中要求的“药品上市后变更不得对药品的安全性、有效性和质量可控性产生不良影响”的要求[13]。此外,若变更与此前生产中重复发生的偏差或稳定性问题相关,则意味着变更前工艺或产品稳定性可能存在风险,那么即使变更后工艺控制能力不降低,变更风险也仍较高。因为若变更没有从根本上解决引起工艺或稳定性研究偏差的问题,那么变更后工艺是否具备持续、稳健生产具有批间一致性产品的能力就需要全面的可比性研究来确证。
除了上述工艺变更的风险,进行工艺变更时还会因变更事项不同,对风险有不同的考量,具体表现如下。
对于生产规模变更,基于调研和实际审评经验,与规模变更风险相关的因素一般包括:①规模的变更是否属于线性变更;②变更是否会增加外源因子风险或无菌控制风险;③变更是否会对产品本身质量及杂质的种类和残留量产生影响;④变更是否会对病毒去除/ 灭活工艺产生影响。
对于工艺程序和步骤变更,若在原液和制剂阶段出现工艺步骤增加、删除或调换,均应按照重大变更管理。对于多组分疫苗、含佐剂疫苗等产品,制剂配置过程中不同成分添加的先后顺序会影响产品关键质量属性,因此针对这些组分加入顺序的变化,也应当按照重大变更管理。
对于生产工艺参数变更,相关风险因素的考量包括:①变更是否会影响病毒去除/ 灭活工艺;②变更是否会对产品质量或杂质种类及残留量产生影响;③变更是否会影响无菌水平/ 微生物限度水平;④对于发酵工艺的变更,还应考虑是否会影响传代代次。一般情况下,放宽、删除、改变关键工艺参数时,变更风险相对较高,且在发生相关变更后,若工艺控制能力降低,则该变更可能不会被批准。
正常情况下,持有人基于既往工艺开发对相关知识的积累,理应对工艺参数进行分类,并建立工艺参数与质量属性的相关性。在此基础上,持有人应参考《变更指导原则》确定合理分级,并开展相应的技术研究。若持有人既往积累的工艺开发知识不足以用来判断工艺参数变化会如何影响相关风险因素,则提示持有人需要开展更多的研究去全面评价变更的影响。因此,即使是同样的变更事项,由于不同持有人对相关知识的积累水平不同、对风险的识别和控制能力不同,相应的,确定变更级别及开展技术研究要求也可能存在差异。
4.4 质量标准的变更
随着国家药品标准的提升、持有人自身产品生产经验的积累、同类产品质量的提升等,生物制品上市后发生标准变更较为常见。鼓励持有人持续提升药品质量标准,药品的注册标准一般应不低于《中国药典》标准,且不低于同类产品的标准,药品标准的变更不允许降低药品的质量控制能力及质量。
一般情况下,删除质量标准的控制项目、替换分析方法、放宽标准限度的变更风险较大,若标准变更会导致质量控制能力降低,这类变更就不符合要求。
如果按照其他关联变更的要求,需要删除内控标准的相关项目,这类变更可能会符合要求。例如,当在生产中不再使用牛血清或抗生素时,持有人将标准中相关残留检定项目删除,这种变更是可以被批准的,或者仅由于方法灵敏度提高,导致可被检出的杂质含量超出已批准范围时,持有人在申请变更检测方法的同时可要求修订标准限度。此外,由于生物制品本身理化性质较为复杂,其产品质量是从工艺和放行不同层面进行控制的,即便增加了标准项目,但若增加的原因是出现了工艺偏差或产品中出现了新杂质等,也可能未从根本上解决问题,变更风险仍较大。
一般情况下,建议持有人优选《中国药典》收录的方法进行检验。若持有人开发的方法更具有产品特异性,为能更好地表征产品质量属性,在将《中国药典》的方法向内部方法变更时,不建议按照中等变更管理。对于检测方法为生物学/ 免疫学/ 免疫化学方法(《中国药典》中的微生物检测除外)的变更,由于这类方法通常用于检验与安全性、有效性相关的关键质量属性,且方法变异度较大,涉及该类方法的变更风险均较大。当发生相关变更时,应由中国食品药品检定研究院对相关变更标准进行单项标准复核和样品检验。若持有人内部对检测方法进行了调整(如调整样品检验时长),且在内部质量体系中经研究、评估认为相关变更对方法学没有影响,不涉及方法学实质性变更且相关变更风险较小,那么这种变更就可在内部质量体系中按照微小变更进行管理。
在变更质量标准时,虽然不涉及工艺变更,不需要开展工艺验证,但持有人应对历史代表性工艺及产品放行、稳定性检测数据进行分析,明确工艺能力和实际产品质量属性变化范围。一方面,持有人可以通过回顾为标准变更提供依据;另一方面,持有人应结合标准变更的内容,分析变更对质量控制能力的影响。对于涉及方法学的变更,持有人应证明拟定分析方法与变更前方法等效或者更优。若质量标准变化会影响稳定性考察,一方面,持有人应采用处于不同考察点的稳定性留样进行考察,用修订后的标准评估稳定性研究结果;另一方面,持有人应承诺以修订后的方法和标准继续进行长期稳定性试验,确保修订后的标准和方法适用于监测产品在贮藏期间稳定性的变化情况,且产品在贮藏期间的质量稳定性应符合变更后的标准限度。
5、良好的沟通交流有助于上市后药学变更管理
由于生物制品本身理化性质复杂,上市后变更情形更是复杂多样,目前我国基于风险的生物制品分类变更经验相对较少,且由于我国实行国家和省级药品监管部门两级监管,良好的沟通对于上市后药学变更管理而言十分重要。
持有人可以根据需要,就变更事项的级别及拟开展的变更研究、先进管理工具的使用等,事先与监管部门进行沟通交流,特别是对于重大变更,持有人应当提前与监管部门就变更风险、变更可比性研究等关键问题进行有效沟通,从而更好地使双方达成共识,避免持有人走弯路,进一步提高研发效率。
6、结 语
生物制品上市后变更较为复杂且广受关注。变更的分类是基于变更的风险进行的,持有人是上市后变更管理的责任主体,需要结合《变更指导原则》的一般要求开展相关研究,并加强与监管部门之间的沟通交流。本文仅针对一般情况对《变更指导原则》进行了进一步的解释、说明,由于变更具有复杂性,持有人还需要具体问题具体分析,结合实际进行变更。