您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-08-02 22:38
午饭期间,和一个器械科室的同事闲聊。
她突然冒出一句:“好羡慕你们啊!平时工作不用耗费很大的脑力”。
小编顿时心生愤愤,我们虽谈不上科学家,但好歹还是个知识工作者。
愤青如小编,特想问一句“为何有此一说?”
那位同事继续说到:“你们检测方法都有标准,直接按照标准做一遍就行了”。
小编突然觉得悲从中来,有一种明珠蒙尘的感觉。
虽然我们做的是仿制药。但是我们的标准真是这样制定的吗?药品杂质是否被全面准确地控制,直接关系到药品的质量可控与安全有效!
怎会像她们说的那么儿戏。所以小编必须写一下我们我们制定标准的过程。今天就先从有关物质开始!
国内杂质检测方法常见问题
身为药学分析研究人员,杂质研究是极其重要内容。常规口服固体制剂,仿制药的药学研究中,杂质研究约占三分之一左右。CDE退审或补充研究,70%与杂质研究有关。
为什么退审或者发补?
绝大多数都是因为踩中了以下几个坑:
1.简单套用药典方法,对自研产品没有针对性,如不知控制何种杂质以及为何控制。
2.方法灵敏度不够。
3.检测方法不可靠,国内外相关杂质调研不充分。
4.缺乏基因杂质,元素杂质的研究。
5.杂质检查方法不适合或方法学验证不完善。
6.杂质检查不可靠,无法准确评价药品质量。
7.未进行检测方法的比较。
如果简单套用已有标准,上述坑必踩。
杂质来源
药品中常见的杂质一般有2个来源。
1.生产过程引入的工艺杂质
在合成药物的生产过程中,可能因为原料不纯或未反应完全、反应的中间体与反应副产物在精制时未能完全除去而引入杂质;
在药物生产过程中,所用的试剂、溶剂、还原剂等可能会残留在产品中而成为杂质。
2.储存过程中的降解杂质
降解杂质主要是在储藏过程中受外界条件的影响,引起药物理化特性发生变化所导致。
在储藏期间因温度、湿度、日光、空气等环境因素的影响,或因微生物的作用,引起药物发生水解、氧化、分解、异构化、晶型转变、聚合、霉变等变化所产生的有关杂质。
有关物质分析方法的建立
所谓分析方法,指分析测试进行的方式。应包含所执行的每个分析试验的必要步骤。如:样品、试剂的制备,参考的比准,使用的仪器等等。
有关物质方法制定依据
1.仿制原料药和制剂相关的杂质谱均不高于原研药
2.掌握各国药典及进口注册标准中原研药API和制剂的杂质谱
所建立的方法中最好可以统筹所有杂质,俗话概括为“套着做”,提高一次通过审评的概率。
例如:XX片,在EP中规定有7个杂质,在进口注册标准中规定有5个杂质,将这些杂质进行结构比对,发现进口标准中的5个杂质中有1个杂质没有被包含在EP中的7个杂质内,那么您所建立的方法应该兼顾这8个杂质(EP中规定的7个杂质和进口标准中没有被包含在EP中的那1个杂质)。
有关物质方法制定要求
1.清晰、详细,且不能产生歧义。
2.所用的分析方法必须具有稳定新指示作用。以便监控原料或制剂在不同存放条件下整个生命周期内杂质的变化情况。
那么问题来了,并且这还是一个“鸡生蛋,蛋生鸡”的问题!
有关物质方法需要在研究之初制定。此时我们尚未获得长期/加速稳定性试验、影响因素实验的情况。如何在不知道杂质的情况下建立分析方法呢?
步骤一:文献检索
查找相关指导原则
如:化学药杂质研究技术指导原则、化学药质量控制分析方法与验证技术指导原则、已有国家标准化学药品研究技术指导原则、ICHQ3A、ICHQ3B、ICHQ3C等。
查找目前国内外该品种杂质谱
查询国内外标准中的杂质谱,经常查询的来源包括:
(1)参考公开信息
(2)药品质量标准(药典、进口标准)
(3)药品说明书
(4)有关文献和专利
(5)咨询从事对照品服务的行业人员等。
检索现行国内外该品种药典原料及制剂检测方法和限度,标准中控制了哪些杂质,并可获知色谱柱类型或是系统适用性图谱。
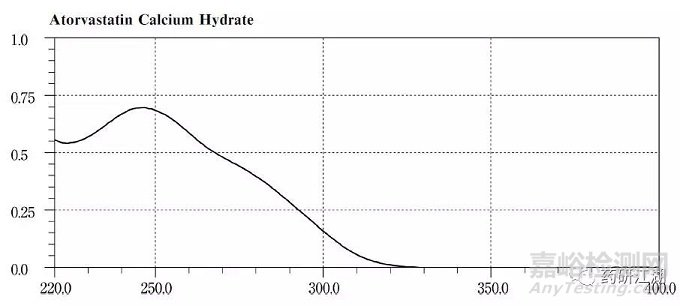
如:阿托伐他汀钙在USP38-NF33中明确查询到色谱柱信息为:

在JP17中查询到的UV图谱为:
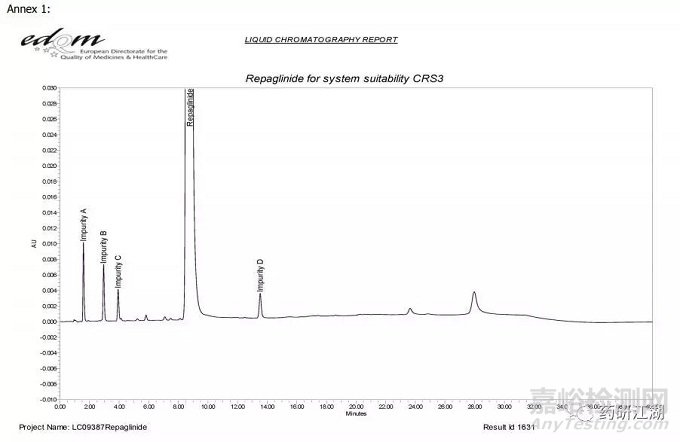
如瑞格列奈在EDQM中记录的系统适用性图谱为:
步骤二:初步试验
一般情况下:
中国药典方法属于最低标准
USP、EP、JP等标准仅收集、整理和评估原研药厂家提供的基本要求,基本没有详细步骤。
进口注册方法可能更接近原研公司内部方法,但可能去掉了关键的细节。
所以应该是利用已查到的药典或标准方法,提高标准制定自研方法,而不是简单套用。方法中包括色谱柱固定相、流动相、洗脱方式、波长、测试样品的处理,具体操作,计算方法等。
1.固定相的选择
首先,试用药典标准或进口注册标准中的色谱柱或效能相当的色谱柱,而后根据研究药物的特性选择合适填料的柱子,以保证:
1.杂质峰与主峰的分离情况达到完美,板数和拖尾因子符合要求
2.确定辅料峰的相对保留时间
3.确定特定杂质的相对保留时间
4.确定有关物质出峰完毕的保留时间
例如:极性化合物的分离可以选用亲水性色谱柱,抗生素聚合物的分离可选用凝胶色谱柱等。
注意不同品牌柱子在耐用性方面的研究。
2.流动相的选择
首先,应该先试用各国药典标准或进口注册标准中的色谱条件,如果分离效果不理想,将色谱条件进行微调(有机相的类型和比例、所用无机盐的浓度和pH值等)。
最重要的是结合杂质的性质和分离情况进行洗脱方式的选择—等度洗脱,梯度洗脱。
类似极性大的化合物可能被掩盖,极性小的化合物难以被洗脱,甚至保留在柱上时,可使用梯度洗脱法。
在分析方法中尤其是转移方案中一定要注明回到初始条件平衡的梯度。
3.波长的选择
在利用DAD检测器进行初步试验测定时,可以选择查看各杂质及主成分的紫外吸收图谱,并记录最大吸收波长,选择最佳测定波长。
波长选择原则:
1.大部分主要杂质与主成分均在此波长下有吸收,可确定此波长为最佳波长。
2.无紫外吸收或检测波长处吸收相差较大的多种化合物,有时在单一检测波长处不能检出,则兼顾各杂质吸收情况,特定波长检测特定杂质,必要时,改用其他类型的检测器。
4.样品浓度
一般情况下,样品浓度在0.1mg/ml-5mg/ml范围内,浓度高时有利于杂质的检出,但需防止超载(浓度超载和进样体积超载)。
例如:XX片中主药的定量限为0.42ng,根据ICH杂质控制的原则,日最大剂量2g,原料的报告阈值为0.05%,原料的报告阈值为0.05%,因此载样量大于0.84μg(0.42ng/0.05%=840ng=0.84μg)即可满足,而供试液的浓度0.5mg/ml,进样量为10ul,载样量为5μg(0.5mg/ml*10μl),完全可以满足测试需要。
5.样品处理
样品的处理不但包括溶剂的选择,还有溶解方式的选择。所选择的溶剂务必要保证主药全部溶解。
溶解方式的表示不要产生歧义。
例如“待XX片全部崩解后,超声使主药溶解”,其中没有具体说明多长时间可以完全崩解,超声需要多长时间等详细信息。应该用回收率试验来验证溶剂对主药的提取程度。发现提取程度不理想,应及时调整样品处理方法。
计算方法
可以遵循药典方法中计算方式并验证方法的合理性。
计算方法一般包括:
(1)峰面积归一化法
(2)杂质对照品法
(3)不加校正因子主成分自身对照法
(4)加校正因子的主成分自身对照法
而对为未知杂质,常用不加校正因子的主成分自身对照或面积归一化法(无法准确定量,无法有效检测杂质)。
在前期科研阶段,并且特定杂质的对照品都可以获得的情况下,最好用杂质对照品法按照外标法来研究,不到可以定位,定量也最准确。然后根据稳定性研究结果确定最后标准时可以结合实际情况考虑订成自身对照法,但是必须提供足够的依据,药学验证也必不可少。
稳定性试验的指示作用
在杂质分析的研究阶段,可以用可能存在的杂质、强制降解产物(光、热、强酸、强碱、强氧化),分别或加入主成分中,配制供试溶液进行色谱分析,调整色谱条件,建立适用性要求,保证方法专属、灵敏。
并且最终分析方法是为稳定性样品服务的,所以建立稳定性指示作用的分析方法是药物质量标准的必须条件。
那么,我们可以在小试阶段设计有意义的破坏试验或具有代表性的降解模式。
例如:XX片在实验过程中发现主药对高温极度敏感,可以将小试样品放置在60℃高温条件下15天左右或是高温高湿(85℃、75%)24小时,查看主药降解杂质。
而所建立的检测方法必须可以灵敏的检测降解杂质的增长趋势。杂质检查与药物外观色泽、含量等项目间存在密切联系,在稳定性放样过程中如果这些项目发生明显变化,而杂质基本不变时,应分析原因,关注杂质检查方法的可行性,必要时重新调整方法,对标准进行修订。
自研方法的验证
在研究时,应牢记质量源于设计的理念,采用几种不同的分离分析方法或不同测试条件以便比对结果,选择较佳的方法作为质量标准的检查方法。
在资料中需列出不同检查方法的比较结果,以证明自研方法可以完全掌握新技术和细节,而自研方法同样需要验证。
验证主要内容包括:
专属性、定量限、检测限、精密度、重复性、中间精密度、线性、准确度、灵敏度、耐用性、样品稳定性等。
有关物质检查方法还要考虑普遍适用性,所用的仪器和试验材料应容易获得。对于特殊试验材料,应在质量标准中写明。验证内容实在是庞大,本文不详述,咱们下次讨论。
有关物质的控制限度
对于药典中规定的特定杂质,其控制限度不得高于药典的限度。如果药典中没有对该杂质设置控制限度,则根据ICH的杂质指导原则Q3A(R)和Q3B(R),同时也参考其它药典来设置该杂质的控制限度。
如果杂质在样品测定中的实际值较高,而需要设置一个高于药典或ICH论证限的控制限度时,则必须提供一个充分合理的论证来说明所设的控制限度是合理的。但将杂质水平降低至药典或ICH论证限以下,是最简单的杂质控制方法。
杂质的合理控制基于多种因素:
患者人群;杂质及含一定限量杂质的药品的毒理学研究结果;给药途径;每日剂量;杂质药理学可能的研究结果;原料药的来源;治疗周期等。
根据ICH的杂质指导原则Q3A(R)和Q3B(R),当满足下述一个或多个条件时,可以认为该杂质的控制限度是合理的:
1.对比参比制剂:当杂质实际观察水平以及控制限度未超出FDA已经批准的人用制剂杂质实际观察水平。
2.杂质是代谢产物:当杂质本身是原料药在动物和/或人体内中药的代谢产物时。(其实该条理论也存在不同看法,有的人认为即使是代谢产物,也应该不超过ICH的论证限)。
3.当杂质实际观察水平以及控制限度有充分合理的科学文献支持时。
4.当杂质实际观察水平以及控制限度未超过通过体外遗传毒性比较研究得到的正确评估限度时。

来源:铭研医药


