您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-09-14 18:55
目前上市销售的化学药品已经超过4000种,许多相似结构药物的药理作用却完全不同。因此,无论是药品生产企业还是药品检验检测机构,根据药品标准进行检验的首要问题是鉴别真伪,以防止由于药品生产和流通的各个环节发生差错而导致药物被误用。
一、HPLC在鉴别中的应用概述
采用HPLC法进行鉴别,除另有规定外,通常是对含量测定项下供试品溶液色谱图中药物主成分峰与相应对照品溶液色谱图中的主成分峰的保留时间进行比较,保留时间应一致。高效液相色谱法中,最常用的检测器为紫外-可见分光检测器,紫外检测器的标准配置为多波长检测器或二极管阵列检测器(简称DAD,有些仪器公司又称之为PDA),采用DAD检测器可对供试品和对照品色谱峰的紫外吸收光谱进行比较,使鉴别更可靠,越来越多的企业开始采用HPLC-DAD法同时进行色谱鉴别和紫外光谱鉴别,即供试品溶液主峰的保留时间及紫外吸收光谱应与对照品溶液主峰的保留时间及紫外光谱一致。
《美国药典》(简称USP)通则<621>指出色谱分析中的保留时间代表了该化合物的一个特征,但不是唯一特性,供试品与对照品保留时间的一致可以用来判定这两种物质是否相同的一个方面,但不足以确定这两种物质的同一性,某些物质的保留时间可能在每张色谱图中都有所差异。各国药典对HPLC法鉴别的规定都是供试品溶液中主峰的保留时间应与对照品溶液主峰的保留时间一致,但都未对一致的程度做出具体规定。如果认为保留时间差异较大,可通过比较色谱峰的紫外光谱以确定两者是否为同一化合物或采用向供试品溶液中加入对照品,应只出现一个主峰的方法来确定是否为同一化合物。
二、HPLC法应用于鉴别时要注意的问题
理论上,同一物质在相同的色谱系统中其色谱行为应完全一致,可在实际工作中通常发现保留时间并不能完全一致,会有一定的波动和漂移。导致波动和漂移的因素有很多,较为常见的主要有以下几种。
(一)柱平衡
流动相组成或温度变化时,色谱柱在新条件下再平衡需要一定的时间,在色谱柱达到完全平衡前不应进样。在反相色谱中,色谱柱很快即可达到平衡,通常用10~20个柱体积的流动相冲洗或运行20~30分钟即可;但在离子对色谱中,一般离子对试剂的浓度都非常低,为使其在色谱柱上达到平衡,需延长平衡时间,极端情况下可能需要几百毫升的流动相方能达到平衡。用硅胶柱作正相分离时常有保留时间突然改变的情况,其原囚是硅胶表面吸附的水含量的影响,流动相发生改变时相应地改变了水的含量,使硅胶柱的含水量发生变化,溶解在流动相中的水对正相色谱的保留时间的影响很大,因为水在正相色谱常用的流动相(如正己烷、二氯甲烷等)中的溶解度非常低,所以色谱柱的平衡通常需要很长时间;如用无水流动相,几乎不可避免地从空气中吸收水分,引起硅胶柱表面吸附水含量的上升,最好的办法是在流动相中加入一定量的水,使色谱柱上水的含量维持恒定。
(二)低浓度强吸附组分
强吸附组分可能永久地键合于色谱柱填料上,或者与填料表面发生化学附着,键合相也有可能被部分地去掉,从而引起保留时间的变化,在此情况下可用保护柱或用强流动相冲洗(反冲更为有效)去掉强吸附组分。低浓度强吸附组分可能由流动相引入,也有可能由样品中带入,后者在药物分析中尤为常见。目前药物制剂的剂型很多,《中国药典》2015年版收载有38种剂型,其中每个剂型又有分类,即使是同品种制剂,不同企业产品中的辅料也不尽相同,因此在鉴别中要考虑辅料对试验可能的影响或干扰。
(三)分离条件发生变化
分离条件的变化主要包括流速、流动相组成、温度等的改变。流速的偶然波动通常是由于泵的问题引起,如存在气泡,但在该情况下,样品间的保留时间比是恒定的;流动相组成的改变可能是由于在线混合泵失灵引起的比例误差,亦有可能是流动相长时间放置后组分发生了改变,后者尤为常见,尤其在使用挥发性溶剂时,真空脱气引起挥发性组分的损失;如色谱柱上未安装恒温装置或安装了恒温装置但未进行控温,在此情况下,当观察到所有色谱峰的保留时间发生连续变化时,可首先考虑环境温度的变化是否引起了保留时间的变化,通常1℃的温度变化可能导致保留时间产生1%~2%的漂移。
(四)其他
除以上几种常见的情况外,由于药品的特殊性,在化学药品的鉴别中还应注意以下一些特殊情况。
复方制剂,由于组分多样性和辅料的添加,有时要采用两种或更多的色谱条件进行药物鉴别。比如USP39中收载的含有17种维生素的复合维生素片剂,需要采用C18、C8、氨基柱和硅胶柱4个色谱系统进行鉴别;对于各组分的紫外响应不同,尤其是含量相差很大的复方制剂,为保证低含量药物的检出,在鉴别中常采用不同的检测波长。比如复方布地奈德/富马酸福莫特罗吸入粉雾剂,富马酸福莫特罗的含量为4.5ug/吸,仅为布地奈德的1/40,布地奈德和富马酸福莫特罗的出峰时间分别约为4分钟和11分钟,根据组分在流动相中的紫外吸收光谱采用不同的波长分别检测布地奈德和富马酸福莫特罗。
除了选择合适的检测器和色谱柱外,样品的处理中,最好采用流动相作为溶剂进行样品制备,对照品和样品的溶剂系统应尽量一致。比如双氢青蒿素,采用乙腈和甲醇为溶剂时,色谱峰完全不同(图14-4)。当然也会有特殊的情况,如盐酸氨溴索注射液有关物质检查,以流动相为溶剂时,杂质B峰面积不断增加,在8小时内,峰面积由1814增加至11 289,而以甲醇作为溶剂时,杂质B则不再增加,此时就不能再采用流动相为溶剂。
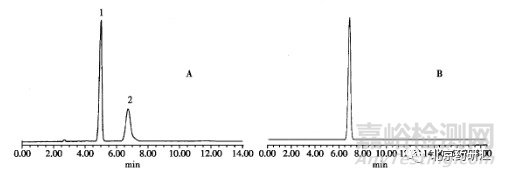
图14-4 双氢青篙素色谱图(A以甲醇为溶剂,B以乙腈为溶剂)
色谱柱:Waters Symmetry Shield RP18150mm×4.6mm
流动相:乙腈-水(1:1)
检测波长:216nm
另外一些药物是与有机酸结合的形式,常见的有机酸包括:枸橼酸、酒石酸、马来酸、甲磺酸、苯磺酸、萘磺酸、琥珀酸等,在采用HPLC法鉴别含有机酸盐的药物时,有机酸盐的鉴别往往被忽略,如果在检测波长下,该有机酸的响应明显,应采用相应的有机酸对照品或确切的对照物质对药品中的有机酸进行鉴别。比如采用苯磺酸和双羟萘酸对照品对苯磺酸氨氯地平、双羟萘酸噻嘧啶中苯磺酸和双羟萘酸色谱峰进行确认(图14-5)。与主成分相比,一般情况下有机酸先出峰,比如双羟萘酸嘧啶、枸橼酸他莫昔芬、苯磺酸氨氯地平等药品。但也不排除有机酸在主成分之后出峰现象,比如苯甲酸利扎曲坦。因此,在标准中应注明出峰顺序、相对保留时间以及有机酸与碱基药物的分离度和峰型。
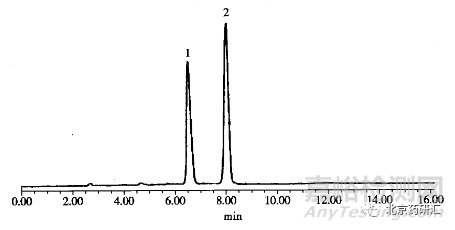
图14-5 双羟萘酸噻嘧啶液相色谱图
1.双羟萘酸;2.噻嘧啶
色谱柱:Waters Symmetry Spheri-10 ODS 250mm×4.6mm
流动相:乙腈-水-乙酸-二乙胺(94:2.5:2.5:1)
检测波长:288nm
三 小结
由于HPLC法鉴别具有准确性高、自动化程度高、样品前处理简单等优点,且通常是在HPLC法含量测定/有关物质项下即可完成,目前已成为各国药典鉴别项的首选方法。

来源:Internet


