您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2019-09-23 17:17
今日,器审中心发布金属髓内钉系统产品注册技术审查指导原则(征求意见稿),全文如下
金属髓内钉系统产品注册技术审查指导原则(征求意见稿)
本指导原则旨在指导和规范金属髓内钉系统产品的注册申报工作,帮助申请人理解和掌握该类产品的原理/机理、结构、主要风险、性能、预期用途等内容,用来指导申请人准备和撰写申报资料。同时也可以用于帮助审评人员把握技术审评工作基本要求和尺度,对产品安全性、有效性做出系统评价。
本指导原则所确定的核心内容是在目前的科技认知水平和现有产品技术基础上形成的。因此,相关人员参考时应注意其适宜性,密切关注适用标准及相关技术的最新进展,考虑产品的更新和变化。随着对产品理解的不断深入,本指导原则相关内容也将适时进行调整。
本指导原则不作为法规强制执行,如有能够满足法规要求的其他方法也可以采用,但应提供详细的研究资料和验证资料。应在遵循相关法规的前提下使用本指导原则。
一、适用范围
本指导原则涵盖的金属髓内钉系统适用于四肢骨折髓内固定,主要包括肱骨髓内钉、股骨髓内钉、胫骨髓内钉、关节融合髓内钉等。金属髓内钉系统通常由髓内钉主钉、锁定部件和封帽组成。通常采用符合GB 23102、GB 4234、GB/T 13810、YY 0605.9等标准中已认可的金属材料制成。
二、技术审查要点
(一)产品名称要求
产品名称应符合《医疗器械通用名称命名规则》。根据产品的结构及组成可命名为金属带锁髓内钉(系统)、交锁髓内钉(系统)等。特征词可包括使用部位、结构特点、植入方式等。
(二)产品的结构和组成
1. 产品结构及组成:金属带锁髓内钉主要由髓内钉主钉、锁定部件和封帽组成。锁定部件可包括螺旋刀片、拉力螺钉、锁定螺钉。
2. 产品型号规格划分:应在注册申报资料综述资料及技术要求中提供产品各型号规格的划分原则。产品的型号划分应考虑产品的设计特点及适用部位,明确各型号的产品描述,包括明确系统结构特点、主钉结构特点、适用部位、骨折部位、植入方式等信息,其中系统结构特点包括交锁、带锁或者不带锁,主钉结构包特点括空心或实心,适用部位包括股骨、胫骨、肱骨等,骨折部位可分为近端、骨干、远端,植入方式分为顺行和逆行。例如股骨顺行髓内钉、股骨逆行髓内钉、股骨干部顺行髓内钉、股骨近端髓内钉等。
应在注册申报资料综述资料中提供主钉的长度、直径、角度(如适用)、锁定孔的结构、尺寸及位置。锁钉的长度、直径。封帽的直径。明确临床使用时各组件间的配合关系。
(三)产品工作原理/作用机理
髓内钉经骨远端或近端置入髓腔内,通过髓内固定的方式达到骨折断端固定,其对骨折的固定为应力分散式固定,而非应力遮挡式固定。
(四)注册单元划分的原则和实例
注册单元划分应符合《注册单元划分指导原则》的要求。
主要组成材料不同的产品原则上应划分为不同的注册单元,作为组合使用的组件可以按同一注册单元申报,如髓内钉主钉、锁定部件(包括:锁钉(或螺栓)、拉力螺钉、螺旋刀片)、封帽等。根据产品的系统结构特点、固定原理不同的产品划分为不同的注册单元,如带锁髓内钉或不带锁髓内钉等。
(五)产品适用的相关标准
包括但不仅限于以下标准:
GB 4234.1-2017 《外科植入物金属材料第1部分:锻造不锈钢》
GB 23102-2008 《外科植入物金属材料Ti-6Al-7Nb合金加工材》
GB/T 12417.1-2008 《无源外科植入物骨接合与关节置换植入物第1部分:骨接合植入物特殊要求》
GB/T 13810-2017 《外科植入物用钛及钛合金加工材》
GB/T 16886.1-2011 《医疗器械生物学评价第1部分:风险管理过程中的评价与试验》
GB/T 16886.5-2017《医疗器械生物学评价第5部分:体外细胞毒性试验》
GB/T 16886.7-2015《医疗器械生物学评价第7部分:环氧乙烷灭菌残留量》
GB/T 19633.1-2015《最终灭菌医疗器械包装第1部分:材料、无菌屏障系统和包装系统的要求》
GB/T 19633.2-2015《最终灭菌医疗器械包装第2部分:成形、密封和装配过程的确认的要求》
YY 0341-2009《骨接合用非有源外科金属植入物通用技术条件》
YY 0605.9-2015 《外科植入物金属材料第9部分:锻造高氮不锈钢》
YY/T 0149-2006《不锈钢医用器械耐腐蚀性能试验方法》
YY/T 0149-2006《不锈钢医用器械耐腐蚀性能试验方法》
YY/T 0316-2016《医疗器械风险管理对医疗器械的应用》
YY /T 0640-2016 《无源外科植入物通用要求》
YY/T 0466.1-2016 《医疗器械用于医疗器械标签、标记和提供信息的符号第1部分:通用要求》
YY/T 0019.1-2011 《外科植入物髓内钉系统第1部分:横截面为三叶形或V形髓内钉》
YY/T 0019.2-2011 《外科植入物髓内钉系统第2部分:髓内针》
YY/T 0591-2011 《骨接合植入物金属带锁髓内钉》
YY/T 0727.1-2009 《外科植入物金属髓内钉系统第1部分:髓内钉》
YY/T 0727.2-2009 《外科植入物金属髓内钉系统第2部分:锁定部件》
YY/T 0727.3-2009 《外科植入物金属髓内钉系统第3部分:连接器械及髓腔扩大器直径的测量》
ISO 5831-1 《外科植入物金属材料第1部分:锻造不锈钢》
ISO 5832-3 《外科植入物- 金属材料- 第3部分:锻造钛6铝4钒合金》
ISO 5832-11《外科植入物- 金属材料- 第3部分:锻造钛6铝7铌合金》
上述标准包括了适用的相关标准。不包括根据产品的特点所引用的其他标准。本指导原则中所列标准适用最新版本。
(六)产品的适用范围/预期用途、禁忌症
产品的适用范围应与申报产品的性能、功能相符,并应与临床评价资料结论一致。
适用范围:该产品通常适用于四肢骨骨折髓内固定。根据申报产品具体的适用部位对适用范围进行细化,如:
股骨:适用于股骨干骨折、股骨颈骨折或合并同侧股骨干、股骨粗隆间骨折或合并同侧股骨干骨折内固定。
肱骨:该产品用于肱骨近端骨、肱骨近端骨折延伸至肱骨干、肱骨近端合并肱骨干骨折的内固定。
胫骨:胫骨干骨折合并移位。
禁忌症:
(七)产品的研究要求
1.产品性能研究
应当提供产品性能研究资料以及产品技术要求的编制说明,包括功能性、安全性指标以及与质量控制相关的其他指标的确定依据,所采用的标准或方法,采用的原因及理论基础。
由于金属带锁髓内钉系统的结构较为复杂,设计验证要求较多,因此本指导原则着重以金属带锁髓内钉为例,介绍相关产品性能研究资料要求,其他髓内钉产品可参照相关要求,选择适用的验证项目进行验证。
申请人应提供产品技术要求的研究和编制说明。对于金属髓内钉系统产品,申请人应结合临床适用部位的解剖结构及置钉方式等,提供关键尺寸参数的确定依据,如髓内钉主钉直径、长度,锁定孔的结构及关键尺寸,锁钉的直径和长度等。
试验用于验证与髓内钉主钉设计和材料有关的弯曲强度和弯曲刚度。申请人可以参考YY/T 0591中的要求开展相关试验。
应关注该方法的适用性,即该方法特指在沿主体长度(WL≥10×直径)明确定义了均匀开放或封闭截面工作长度(WL)的带锁髓内钉,并且其应用于股骨、胫骨、肱骨等骨干全长。
髓内钉扭转刚度会对周围骨和骨痂内的载荷转移和应力水平有影响,并且会影响骨愈合的速率和强度以及长期的改建。髓内钉静态扭转试验用于髓内钉性能的比较。
在骨支撑不足并且延迟愈合的案例中,髓内钉的循环弯曲强度会变得很重要。在这些案例中,在骨愈合并产生足够的力学支撑来降低髓内钉的应力水平之前的几周时间内,髓内钉工作长度的非支撑区域可能会产生主应力。髓内钉弯曲疲劳试验用于确定髓内钉循环弯曲强度。
在植入人体后,锁钉会受循环弯曲力作用,锁钉的循环弯曲强度会变得很重要。锁钉弯曲疲劳试验用于确定锁钉循环弯曲强度。
由于髓内钉系统在人体中的受力方式及受力水平未知,会受到多种因素如病人活动的程度和类型、周围骨和软组织的状态、骨折类型、病人体重等的影响,而YY 0591中给出的各种力学性能研究方法并未考虑上述因素,因此申请人很难根据实验结果预判产品临床实际风险,应当结合产品适用部位和结构设计以及骨折分型等,设计相关力学性能论证产品设计、尺寸以及材料等性能的合理性。
对于带锁髓内钉产品用于股骨近端骨折(如转子间骨折、转子下骨折),需选取结构设计的最差情况,评价拉力螺钉/螺旋刀片/重建锁定螺钉与髓内钉主钉(下称主钉)连接部位的结构疲劳性能。通过包埋介质将主钉固定在试验夹具内,夹具内表面带有防止包埋介质旋转的凹槽设计,使用软塞将主钉远端锁定孔和中空开孔封堵,防止包埋介质填充。主钉的远端干部(内外方向)应在夹具的垂直平面内,主钉近端外偏角度模拟临床使用情况,根据骨折断面位置的最差情况设置主钉与拉力螺钉/螺旋刀片/重建锁定螺钉的连接部位与包埋平面的距离。按照制造商推荐的临床使用方法将螺钉置入股骨头(聚氨酯泡沫或其他模量和强度合适的高分子聚合物)中,根据预设的股骨头球心与主钉轴线的水平方向距离,将拉力螺钉/螺旋刀片/重建锁定螺钉与主钉锁定。将夹具固定在试验机基座上,倾斜角度可参照ISO 7206-4: 2010标准中规定的内收角度α(10°±1°)和后倾角度β(9°±1°)。确定106次循环次数的疲劳极限,并结合临床使用部位的受力情况,说明其接受依据。
对于带锁髓内钉产品用于股骨、胫骨远端干部骨折,需选取结构设计的最差情况,评价远端锁定螺钉与主钉的连接部位的结构疲劳性能。将主钉远端固定在试验夹具或模拟骨制成的股骨内模型内,根据临床使用推荐的工作距离最差情况设置骨折断面与最靠近近端的远端锁定孔的距离,置入远端锁定螺钉。按照推荐的临床使用方法将近端螺钉置入股骨头(聚氨酯泡沫或其他模量和强度合适的高分子聚合物)中,将螺钉与髓内钉锁定。将夹具固定在试验机基座上,倾斜角度可参照ISO 7206-4: 2010标准中规定的内收角度α和后倾角度β。确定106次循环次数的疲劳极限,并结合临床使用部位的受力情况,说明其接受依据。
结合产品使用部位、结构设计等选择最差情况进行力学性能研究。最差情形可以借助有限元分析等手段选取。
YY/T 0591中规定的静态四点弯曲性能、髓内钉静态扭转试验、髓内钉弯曲疲劳试验、锁定螺钉弯曲疲劳强度试验等试验用于对比产品设计、材料及尺寸等性能,建议选取最细的髓内钉和锁定部件进行相关试验。如有其他情形,申请人应提供最差情形选择的合理依据。
c.试验结果接受条件的确定依据
试验建议选用对比法,对比试样可选用已在中国境内上市使用的同类器械,产品的材料、性能、结构、组成及适用范围应具有可比性。
对于钛合金产品,若其表面进行阳极氧化处理,为了评价表面处理工艺可能对产品性能产生的影响,可按照YY/T 1615《外科植入物钛及钛合金阳极氧化膜通用要求》对材料的基体和阳极氧化层进行表征。
①锁定部件的切出性能(Cut out)研究
申请人需考虑拉力螺钉/螺旋刀片的设计(如螺纹/螺旋结构设计、组件间的连接结构设计、尺寸规格等)、适用的骨折分型等因素,提供拉力螺钉/螺旋刀片在体外模拟条件下抵抗移位和切出的性能研究资料。试验方法可参照附录1。
②作为组合使用的组件材料不相同的,必要时应考虑电偶腐蚀效应。
YY/T 0591 中要求制造金属带锁髓内钉锁定部件的材料应与主钉材料一致。因此制造商若采用不同金属材质的锁钉部件与主钉配合,则需综合考虑材料的种类、产品植入时间等内容,必要时开展电偶腐蚀性能的研究。
产品若预期在MRI环境中使用,建议开展MRI兼容性测试,否则应在说明书中予以警示。
2.生物相容性评价研究
申报产品的生物相容性评价,按照GB/T 16886.1-2011《医疗器械生物学评价第1部分:风险管理过程中的评价与试验》中的系统方法框图及《国家食品药品监督管理局关于印发医疗器械生物学评价和审查指南的通知》(国食药监械〔2007〕345号)中的审查要点进行风险评价,在缺乏相关数据时,应进行必要的生物相容性试验。
3.灭菌工艺研究
对于经辐照灭菌的产品,需明确辐照剂量及相关的验证报告,具体的剂量确定依据可参照GB 18280系列标准。
对于经环氧乙烷灭菌的产品,需提供灭菌结果确认和过程控制报告,具体可参照GB 18279系列标准。
4.产品有效期和包装研究
(1)申请人应提交产品有效期研究确认资料,依据《无菌医疗器械包装试验方法第1部分:加速老化试验指南》(YY/T 0681.1)对产品(包括包装)采用加速老化试验和实时老化试验的方式验证其有效期。产品的实时稳定性试验和加速稳定性试验应同时进行。如果首次注册时提交的是加速老化验证资料,申请人应继续实时稳定性试验。当加速老化试验结果与其不一致时,应以实时稳定性试验结果为准。
(2)申请人应提交包装研究资料,依据有关国内、国外标准(如GB/T 19633.1、GB/T 19633.2、ISO 11607、ASTM F2475、ASTM D4169等)对包装进行分析研究和评价。直接接触产品的包装材料的选择应至少考虑以下因素:包装材料的物理化学性能;包装材料的毒理学特性;包装材料与产品的适应性;包装材料与成型和密封过程的适应性;包装材料与灭菌过程的适应性;包装材料所能提供的物理、化学和微生物屏障保护;包装材料与使用者使用时的要求(如无菌开启)的适应性;包装材料与标签系统的适应性;包装材料与贮存运输过程的适应性。
(3)根据《无源植入性医疗器械货架有效期注册申报资料指导原则(2017年修订版)》的要求,无论加速稳定性试验还是实时稳定性试验,注册申请人均需在试验方案中设定检测项目、检测方法及判定标准。检测项目包括产品自身性能检测和包装系统性能检测两方面。前者需选择与医疗器械货架有效期密切相关的物理、化学检测项目,涉及产品生物相容性可能发生改变的医疗器械,需进行生物学评价。如适用,可采用包装封口完整性检测用于替代无菌检测。后者则包括包装完整性、包装强度和微生物屏障性能等检测项目。其中,包装完整性检测项目包括染色液穿透法测定透气包装的密封泄漏试验、目力检测和气泡法测定软性包装泄漏试验等;包装强度测试项目包括软性屏障材料密封强度试验、无约束包装抗内压破坏试验和模拟运输试验等。
(八)产品的主要风险
需根据YY/T 0316《医疗器械风险管理对医疗器械的应用》,充分识别金属髓内钉系统产品的设计、原材料、生产加工、包装、灭菌、运输、贮存、使用等全生命周期内各个环节的安全特征,从能量危害(若涉及)、生物学危害、环境危害、有关使用的危害、因功能失效、老化及存储不当引起的危害等方面,对产品进行全面的风险分析,并详述所采取的风险控制措施。
根据金属髓内钉系统的临床应用及发展史,其主要风险包括(但不限于):
髓内钉相关设计的风险包括:结构不合理导致的断裂、生物相容性、感染、固定失效等;
髓内钉生产的风险主要包括:错误材料的使用、错误部件标记、标签错误、灭菌包装的密封性、灭菌过程等。
已识别或可预见导致髓内钉临床使用风险包括:将髓内钉用于禁忌症或不适宜的患者;大号螺钉太强的压力;过度使用切削工具,钝的切削工具的使用;重复使用一次性器械等。
根据金属髓内钉系统的主要结构特点及预期用途,产品的技术指标主要包括产品的外观、尺寸、物理性能、化学性能和无菌性能等。不同申请人的产品参数根据设计要求可有所区别,并根据自身产品的技术特点制订性能指标要求,但不得低于相关强制性国家标准、行业标准的要求。具体内容可参见YY 0341标准。
主要性能指标包括:
主钉的长度、直径;锁钉的直径。
包括主钉和锁钉部件的配合性能及主钉和封帽间的配合性能。
可参考《YY/T 1615-2018 外科植入物钛及钛合金阳极氧化膜通用要求》在技术要求中规定相关性能指标。
按照上述要求,主钉、锁定部件以及封帽等组件应从上述性能中根据其适应性分别制定相关性能指标要求。
(十)同一注册单元内检验典型性产品确定原则和实例
髓内钉主钉、锁定部件和封帽均应分别进行检测。锁定部件中拉力螺钉、螺旋刀片、骨栓和锁定螺钉应分别进行检测。如检测的样品不是所申报的所有产品,则应提供典型性样品的确定依据。
(十一)产品生产制造相关的要求
1. 根据产品设计方案,明确产品工艺流程,要求企业提交工艺流程图,标明产品关键工序和过程控制点。如:产品的主要工艺有机加工、折弯成型、清洗、包装、灭菌等工序。
需提供折弯成型、钛合金产品阳极氧化(如涉及)、清洗和精洗以及灭菌等关键工艺和特殊工序的工艺验证资料。
2. 应要求企业提交研制、生产场地的相关信息,如:场地平面图。如有多个研制、生产场地,应当概述每个研制、生产场地的实际情况。
(十二)产品临床评价要求
注册申请人需按照《医疗器械临床评价技术指导原则》(国家食品药品监督管理总局通告2015年第14号附件)的要求选择合适的临床评价路径提交临床评价资料。
1. 同品种医疗器械评价路径
在满足注册法规要求的前提下,可按照《医疗器械临床评价技术指导原则》进行同品种产品的临床数据对比、分析、评价,并按照《医疗器械临床评价技术指导原则》要求的项目和格式出具评价报告。
选择通过同品种医疗器械临床试验或临床使用获得的数据进行分析评价,需首先将申报产品与一个或多个同品种医疗器械进行对比,证明二者之间基本等同。与同品种医疗器械进行对比的内容包括定性和定量数据、验证和确认结果,应详述二者的相同性和差异性,对差异性是否对产品的安全有效性产生不利影响,应通过申报产品自身的数据进行验证和/或确认,如申报产品的非临床研究数据、临床文献数据、临床经验数据、针对差异性在中国境内开展的临床试验的数据,或采用符合要求的海外临床试验数据。
2. 临床试验评价路径
选择进行临床试验,则应严格按照《医疗器械临床试验质量管理规范》进行临床试验,并提交完整的临床试验资料。
开展临床试验研究时,在临床试验方案制定中需要考虑的因素:
(1)临床试验设计类型
建议采用具有良好对照的前瞻性的随机对照临床试验。
对于常规设计的金属髓内钉系统,鉴于该类产品在实际临床应用中表现良好,且在研发及生产过程中没有发生本质改变,仅在结构设计、表面处理等方面进行改进或仅仅进行仿制,可在临床试验设计中应用单组目标值法,即临床试验不设立对照组。
(2)临床试验观察时间
临床试验的持续时间取决于所有安全性和有效性数据的获得,应随访至骨折的临床愈合,至少为6个月,也可根据实际研究内容,适当延长观察时间。应在术后7天内、6周、12周、24周进行临床随访评估。
(3)临床试验评价指标及判定标准
a.主要评价指标
主要评价指标以“产品有效率”为主要评价指标,将“产品有效”定义为同时满足以下要求(a)骨折愈合评价标准:局部无压痛及纵向叩击痛,局部无异常活动;术后24周骨折部位正侧位X线片上骨折间隙模糊或消失,或者正侧位X线片上可见连续性骨痂越过骨折线;(b)术后24周受试产品无变形或断裂。
b.次要评价指标
有效性指标:(a)手术复位内固定后骨折愈合时间;(b)手术医生对产品术中操作性能的评价,如手术时间,切口长度,术中出血量等;
此外,对于关节周围骨折,可纳入功能评分评价产品的有效性:
i.肱骨骨折采用constant-murley肩关节功能评分;
ii. 股骨转子间骨折采用Harris髋关节功能评分;
iii. 股骨远端骨折采用AKS膝关节功能评分;
iv. 胫骨远端骨折采用Mazur踝关节功能评分。
安全性指标:随访期间应观察金属髓内钉主钉和锁定部件的变形、断裂、松动,以及骨折不愈合、延迟愈合等不良事件的发生情况。
(4)病例选择
a.入选标准
①年龄范围:一般为18-80岁的患者;
②性别;
③疾病原因:创伤骨折、骨折后畸形愈合或不愈合。
④骨折分型;
b.排除标准
如骨髓炎;病理性骨折;骨折伴有严重的软组织损伤;骨折合并血管损伤;骨折合并骨筋膜室综合征;严重骨缺损;严重多发伤;研究者通过病史与骨折部位X线片判断存在严重骨质疏松;全身系统性疾病;使用化疗药物;接受放射治疗;系统性使用皮质类固醇激素;使用生长因子;长期使用镇静催眠药(连续使用3个月以上);长期使用非甾体类消炎药(连续使用3个月以上);一年内有酗酒;不能保证在骨折愈合期间戒烟;药物滥用;研究者判断不适合入选的其他情况(如:代谢性骨病、小儿麻痹后遗症等);拒绝签署知情同意书等。
c.受试者退出标准及退出受试者的处理
退出标准
①受试者撤回知情同意书;
②研究者认为不再适合继续进行临床试验者;
③受试者死亡;
④受试者失访;
⑤申办者要求终止试验。
退出受试者的处理
①记录最后一次生命体征、术后情况和局部体征检查、影像学资料和不良事件等;
②将终止试验的时间和原因详细记录在病例报告表上;
③对因不良事件而终止试验的受试者必须随访至不良事件得到解决或稳定;
④医疗器械临床试验质量管理规范规定的其他相关事宜。
d.不同部位骨折病例的纳入比例
由于上肢和下肢的受力方式、愈合机理以及手术方式的不同,建议合理分配上下肢病例的纳入比例,下肢占比不应低于50%。
(5)对照产品的选择(如适用)
若选择随机对照试验,对照产品应选择目前临床正广泛使用的、对相应适应证的疗效已被证实并得到公认的来源于同一厂家生产的同一系统产品。对照产品的材料、设计、适应证与试验产品具有可比性。应提供对照产品的选择依据。
(6)样本量的估算
申请人应提供样本量足以评价该类产品安全性和有效性的统计学依据,包括以下内容:同类产品临床认可的主要评价指标的目标值、受试产品主要评价指标的预期疗效、I型误差α、Ⅱ型误差β;所用到的样本量计算公式;失访率的合理估计;使用的统计软件;引用的参考文献等。若进行随机对照非劣效试验,则需明确对照产品预期疗效和临床认可的非劣效界值;申请人应根据各自产品的性能指标选择对照品,并采用经典的统计学方法及国内外公认的统计学软件计算样本量。
(7)人口统计学和基线特征
a.人口统计学资料:如性别、年龄、民族、身高、体重等;
b.临床疗效相关的基线数据:考虑因素包括疾病的阶段和程度、临床分类、疾病亚组,如骨折原因、骨折后至手术时间、骨折的临床症状观察和局部体征检查、骨折部位正侧位X线检查,软组织损伤类型(开放性/闭合性)、AO分型(组、亚组、限定)等;
c.既往病史:是否有骨质疏松、营养不良(钙、磷、蛋白质、铁等)、贫血、激素缺乏(生长激素、甲状旁腺素等)、放射治疗、吸烟、嗜酒、手术史、糖尿病史等。
(8)统计分析方法
数据分析时应考虑数据的完整性。所有签署知情同意书并使用了受试产品的受试者必须纳入最终的统计分析。数据的剔除或偏倚数据的处理必须有科学依据,并在研究方案中预先说明。
临床试验的数据分析应基于不同的分析集,通常包括全分析集、符合方案集和安全集,研究方案中应明确各分析集的定义。全分析集中脱落病例主要评价指标缺失值的填补方法(如最差值法等)应在方案中予以说明,并进行灵敏度分析,以评价缺失数据对研究结果稳定性的影响。主要评价指标的分析应同时在全分析集和符合方案集上进行;安全性指标的分析应基于安全集。
应在方案中预先写明具体的统计分析方法、统计分析软件及版本。
计算主要评价指标的95%置信区间。当受试产品主要评价指标的95%置信区间下限大于目标值,认为该临床验证成功,即受试产品达到行业认可的目标。若进行随机对照试验,受试产品与对照产品主要评价指标差值的95%置信区间下限大于非劣效界值,认为该临床验证成功。
对于同一患者存在两处骨折的情况,应对试验结果进行灵敏度分析,即分别对病例(可以随机选择一处骨折,或选择两处骨折中疗效较差者)及例次(所有植入物)进行统计分析。
对验证期间发生的所有有害事件的种类、严重程度、发生频率及与验证产品的关系应列表描述。
3.关于接受境外临床试验数据
对于已有境外临床试验数据的医疗器械,其临床试验如符合《接受医疗器械境外临床试验数据技术指导原则》中的情形,可按照该路径提交临床评价资料,以论证产品的安全性和有效性。
(十三)产品的不良事件历史记录
不良事件的原因包含:感染、过敏反应、周围组织激惹、断裂、固定失效、内植入变形等。
召回原因涉及包装、标志及标签错误、产品加工错误等原因,相关单位及部门可引以为戒,加强生产加工流程管控,减少错误的发生。
(十四)产品说明书和标签要求
金属髓内钉系统的说明书和标签应符合《医疗器械说明书和标签管理规定》(国家食品药品监督管理总局局令第6号)和YY/T 0466.1中的相关要求。说明书、标签的内容应当真实、完整、科学,并与产品特性相一致,文字内容必须使用中文,可以附加其他语种。说明书、标签中的文字、符号、图形、表格、数据等应相互一致,并符合相关标准和规范要求。
产品临床适用范围/适应证、禁忌证、注意事项应依据临床评价或临床试验的结果进行确定。
产品有效期、灭菌产品采用的灭菌方法、非灭菌产品推荐采用的灭菌方法等信息应与产品技术报告所述一致。
说明书的警示中注明MRI内容,明确相关的试验结果,提示其存在的风险。
附录1
拉力螺钉/螺旋刀片切出试验(Cutout Test)
目前,股骨带锁髓内钉用于转子间骨折内固定时,存在股骨头轴向或侧向移位和拉力螺钉或螺旋刀片从股骨头切出(cutout)失效的风险[1]。固定失效的影响因素包括骨折类型和不稳定性(如内侧壁是否完整)、解剖复位的质量、骨质疏松程度以及拉力螺钉或螺旋刀片放置位置(尖顶距离TAD,Tip-Apex Distance)[2-3]。本试验方法综合多种临床使用中的不利因素,如髋关节受力、骨质疏松、不稳定骨折类型和不理想的置钉位置等,评价不同设计的拉力螺钉/螺旋刀片在体外模拟条件下抵抗移位和切出的性能。
采用不超过15pfc等级的聚氨酯泡沫(ASTM F1839)模拟骨质疏松的股骨头,聚氨酯泡沫制作成圆柱状,股骨头直径50mm。按照制造商推荐的手术方法将拉力螺钉或螺旋刀片置入股骨头中,然后将股骨头置入不锈钢套筒中(厚度5mm),尖顶距离TAD和螺钉的轴向偏距可参照文献中的参数设置(尖顶距离TAD为12.2mm,螺钉长轴与股骨颈轴线平行且向后偏心7mm[4]或尖顶距离TAD为45mm,螺钉沿股骨颈轴线置入,股骨颈轴线与不锈钢套筒的轴线平行且向下偏心5mm[6])。
将拉力螺钉/螺旋刀片固定在基座夹具上,螺钉与夹具水平面的夹角保持与主钉在冠状面上角度一致,固定方式与螺钉与主钉的连接方式和约束相同。不锈钢背板(厚度5mm)与套筒机械固定,与聚乙烯支撑结构接触,模拟拉力螺钉/螺旋刀片滑动后不稳定转子间骨折(后内侧壁不完整)断面的接触的情形,允许股骨头内翻和旋转,但是限制股骨头向外侧移动。加载方向通过股骨头球心垂直向下,加载方向的倾斜角度可参照文献设定(夹角为19°,关节合力矢量角度16°加上与股骨解剖轴外翻角度3°[4];或夹角为23°,关节合力矢量角度18°加上与股骨解剖轴外翻角度5°[5];或加载方向与螺钉轴线的夹角29°[6])。通过水平轴承和聚乙烯球窝衬垫对股骨头施加循环压缩载荷,直至达到规定的循环周期或螺钉切出。加载方式可参照文献设定(正弦曲线,R=0.1,频率3Hz,循环周期100000,载荷水平为0.8kN、1.0kN、1.2kN、1.4kN[4];或峰值为1.45kN的双峰曲线[5];或加载方向同时考虑髋关节屈伸角度,阶梯式加载,初始载荷600N,每个阶段700次循环,下一阶段载荷增加50N,直至螺钉切出失效[6])。试验装置图如图1所示。
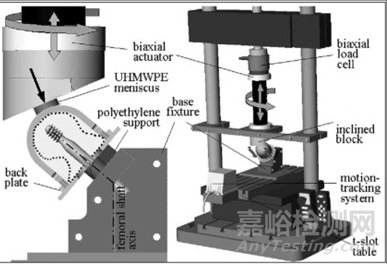
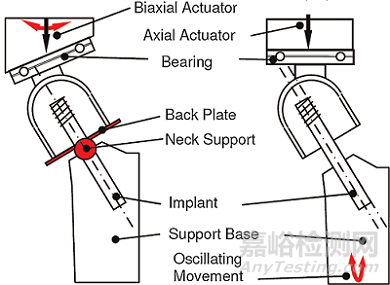
图1 试验装置图
通过测试螺钉与不锈钢套筒之间的导电性探测螺钉是否发生切出,当螺钉切出股骨头与不锈钢套筒接触时,触发控制器停止试验机加载,记录经历的循环周期NCO。另外,通过三维运动捕捉系统记录股骨头相对于螺钉的三维位移数据,计算股骨头内翻角度αvarus和绕股骨颈轴旋转角度αneck。
参考文献
[1] Yoshimine F, Latta LL, Milne EL. Sliding Characteristics of Compression Hip Screws in the Intertrochanteric Fracture: A Clinical Study. J Orthop Trauma. 1993;7(4):348-53.
[2] Kim W, Han C, Park J, et al. 2001. Failure of intertrochanteric fracture fixation with a dynamic hip screw in relation to pre-operative fracture stability and osteoporosis.IntOrthop 25:360–362.
[3] Baumgaertner MR, Curtin SL, Lindskog DM, et al. 1995. The value of the tip apex distance in predicting failure of fixation of peritrochanteric fractures of the hip. J Bone Joint Surg Am 77:1058–1064.
[4] Sommers MB, Roth C, Hall H, et al. 2004. A laboratory model to evaluate cut-out resistance of implants for pertrochanteric fracture fixation. J Orthop Trauma 18:361–368.
[5] Ehmke LW, Fitzpatrick DC, Krieg JC, et al. 2005. Lag screws for hip fracture fixation: evaluation of migration resistance under simulated walking. J Orthop Res 23:1329– 1335.
[6] Christopher T. Born, Bernhard Karich, Christoph Bauer, Geert von Oldenburg, Peter Augat. Hip Screw Migration Testing: First Results for Hip Screws and Helical Blades Utilizing a New Oscillating Test Method. J Orthop Res. 2011 May;29(5):760-6.

来源:中国器审


