您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2019-06-12 14:57
对原料药工艺验证的一些问题作了个人的探讨。对工艺验证的由来、定义、原因以及工艺验证的三个阶段作了简单的讨论与阐述。希望有更多的实际案例学习。
“验证”一词,出自东汉王充的《论衡·奇怪》:“言之有头足,故人信其说;明事以验证,故人然其文。”意思是:说话有道理,所以人们相信他的说法;事情可以验证,所以人们认可他的文章。这充分体现了唯物主义思想精神,所谓原料药的工艺验证,就是为了用“验证结果”的客观事实来证明“设计工艺”的真实可靠性。下面谈一谈本人对工艺验证内容的一些理解。
关于工艺验证的具体详细内容,可以阅览“药品生产验证指南”、ICH Q7、FDA颁布的关于工艺验证的相关指导文件。里面有详细的指导与介绍。下文只对一些个人的理解作下阐述,与大家共同探讨。
一、验证的由来
1970年至1976年,美国爆发了一系列由于受污染的输液导致的败血症病例。仅1971年的3月份,就新增405例败血症患者。
调查经历了几年时间。调查结果表明,与败血症案例相关的批次并不是由于企业没做无菌检查或违反药事法规,而在于无菌检查本身的局限性、设备或系统设计建造的缺陷以及生产过程中的各种偏差及问题。FDA从调查的事实看出,输液产品的污染与各种因素有关,如厂房、空调净化系统、水系统、生产设备、工艺等。
FDA当时认为有必要制定一个新的文件,以“通过验证确立控制生产过程的运行标准,通过对已验证状态的监控,控制整个工艺过程,确保质量”为指导思想。这个文件即1976年6月1日发布的“大容量注射剂GMP规章(草案)”,它首次将验证以文件形式载入GMP史册。
二、什么是工艺验证?
在开始探讨工艺验证之前,首先我们得搞明白一个问题,那就是什么是“工艺验证”?这是基本,因为本人发现,有些资料上对工艺验证描述的内容,到后来就变成一种为了申报资料,完成套路而为。一些论坛区,有些关于工艺验证方案的问题已经明显不合法规要求,却仍希望得到肯定支持的答复,自己骗自己,掩耳盗铃。引用黎巴嫩著名诗人纪伯伦的话:我们已经走得太远,以至于我们忘记了为什么而出发。
工艺验证的定义
|
中国GMP |
工艺验证应当证明一个生产工艺按照规定的工艺参数能够持续生产出符合预定用途和注册要求的产品 |
|
欧盟EU |
The documented evidence that the process, operated within established parameters, can perform effectively and reproducibly to produce a medicinal product meeting its predetermined specifications and quality attributes |
|
为证明工艺在设定参数范围内能有效稳定地运行并生产出符合预定质量标准和质量特性药品的验证活动 |
|
|
美国FDA |
Process validation is defined as the collection and evaluation of data, from the process design stage through commercial production, which establishes scientific evidence that a process is capable of consistently delivering quality product |
|
收集和评估从工艺设计研发到商业化生产阶段的数据,并用这些数据科学的证明该工艺能够有能力持续稳定地生产出优质产品 |
其中美国FDA的定义,似乎更为科学,因为其包含了工艺设计开发、工艺验证以及商业化生产后期的持续工艺验证过程。工艺验证不单指申报注册中的三批工艺验证批次的生产(一般生产商考虑成本等问题,都是做连续三批验证工作),而是原料药从开发到生产的整个生命周期。是为保证稳定持续地生产合格产品。
三、为什么要工艺验证?
可能有的人第一反应估计是:因为申报资料需要,不做不给批。
其实工艺验证的主要目的,定义里已经明确说明了:为了持续稳定地生产出符合质量预期的药品。这其实是为了生产商自己的利益考虑,否则一旦出现原料药质量问题,不仅患者的利益收到损害,企业更是会面临倒闭的风险。远的例子不说,最近的案例如长春生物制药,私改设备规模,却没有进行验证,直接导致产品质量不合格,停产退市。所以说,原料药工艺验证不仅是为了患者用药安全,更是为了药厂自己。
药物在得到许可批准上市之前,其原料药的生产工艺必须验证。原料药和关键中间体需在cGMP条件下生产。墨菲定律说道:“可能会出错的事,就一定会发生”。这运用在原料药生产中其实比较合适。为了使生产的出错率降低至最低,我们就需要考虑到各个环节,对可能存在的问题进行研究与验证。总结一句话:工艺验证是为了持续稳定的得到合格产品。
因此,当工艺验证遇到问题时,还是老老实实研究分析,别想能蒙混过关,否则以后的代价可能会更大,除非你有后悔药。
四、什么时候要进行工艺验证?
简单两点:
1、要开始进行一个新产品的生产时,需要进行工艺验证。
2、各种变更。如工艺发生变更;批量发生变更;生产场地发生变更等;起始物料发生变更等。
关于变更的补充申请中,可以分为三类。二类和三类才需要进行相应的验证工作:
第一类:微小变更,对产品质量不产生影响。
第二类:中度变更,须通过相应的研究验证工作证明对产品的质量没有产生影响。
第三类:重大变更,须通过系列的研究验证工作证明对产品的质量没有产生负面影响。
五、怎行进行工艺验证?
工艺验证过程中需要做的工作
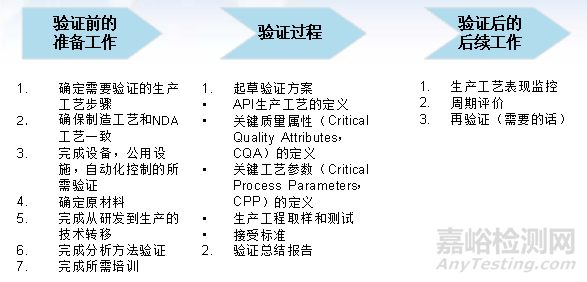
上表中基本概述了原料药整个工艺验证过程中需要做的工作。每一项如果具体讲,都是相当复杂的内容。这也说明了,原料药的工艺验证是一项系统性、复杂性和科学性的工作。需要文件化、制度化来认真对待每一个环节。
1)第一阶段:工艺研发
俗话说的好:巧妇难为无米之炊。如果没有已经仔细研究的初步生产工艺,任凭设备如何先进,GMP管理规章制度实施的有多好,都是不可能完成工艺验证的。
工艺研发是工艺验证的起点。关键质量属性(CQA)和关键工艺参数(CPP)需要在该过程中完成初步研究及确定。
关于CQA和CPP的定义为:
CQA (Critical Quality Attribute):产品的物理,化学,生物或微生物性质或特征,应在适当的限度,范围或分步之内,以确保预期的产品质量
CPP (Critical Process Parameter):指其波动会影响到关键质量属性的工艺参数,应该被监控或控制,以确保工艺能生产出预期质量的产品
关于这方面之前有些疑惑:关键质量属性和药典质量标准有什么区别?或者说,质量标准中,哪些因素不属于关键质量属性呢?
一般较为官方或文献答案,指出:关键质量属性不等同于药典质量标准。这好理解,因为我们自身的生产工艺与药典中原料药的生产工艺未必相同。比如药典中残留溶剂控制了二氯甲烷,而我们的原料药整个过程中,从未使用二氯甲烷,最终精制过程中使用了乙酸乙酯。那么二氯甲烷的残留肯定不属于关键质量属性,乙酸乙酯的残留量才属于关键质量属性。
确定关键质量属性,是为了避免研发人员仿标准而不是仿质量。
假如一种情况,自身的生产工艺与药典的工艺一致,那药典标准中的所有指标是否都是关键工艺参数呢?本人觉得是的。虽然说这与客户的具体需求有关,但你很难说哪个是不关键的。因为如果不关键,那标准中也根本无需提出来。比如性状,药典指出是白色粉末,而你生产出了绿色粉末,即使其他检查项都百分百合格,这个API有人会要吗?
还有一个问题,哪些是关键工艺参数?因为,对有些原料药的工艺,有人觉得,很简单啊,没什么关键工艺参数的。或者是,觉得所有参数都很重要啊,哪个参数的改变都会带来质量变化。这时,解决方法似乎很符合官方的回答方式:你需要引入风险评估。确定关键工艺参数,是为了避免研发人员对产品质量只是通过终产品检测,而不是质量源于设计的思路。
其实,CQA和CPP的引入,个人觉得完全就是为了通过这些参数的确定,让研究者对工艺的深入理解,从而保证合格产品的稳定与持续生产。
关于工艺验证实施前和操作规程准备的一些考虑
|
确定目标 |
质量标准、投料量、时间节点、客户的相关需求 |
|
安全 |
公斤级实验需要完成风险评估,中式和生产强制执行风险评估 |
|
背景回顾 |
与之前的工艺开发者进行交流 |
|
确定关键步骤 |
加料,反应,后处理,浓度,结晶,分离,干燥 |
|
设备限制 |
低温能力,防腐能力,离心能力 |
|
明确的过程控制 |
取样,分离,定量 |
|
反应未完全的应急策略 |
延长反应时间,增加反应试剂,继续往后做反应 |
|
失控反应的应急策略 |
缓冲槽,安全设备 |
|
延伸操作实验的结果预计 |
在小试实验室进行 |
|
破环实验的结果预计 |
在小试实验室进行 |
|
产物分析方法的开发 |
优选简单的分析方法 |
|
确证工艺的耐受性 |
溶剂±5ol%,试剂±wt% |
|
确认清洁操作 |
一般基于怎样能溶解产物 |
|
确认三废处理方法 |
中和,分解 |
2)第二阶段:工艺验证
工艺验证方案构成要素,这些要素都需要实行文件化管理
|
验证范围 |
关键工序 |
|
验证职责 |
工序试验 |
|
工艺描述 |
取样计划 |
|
物料 |
接受标准 |
|
设备 |
偏差与调查 |
|
厂房 |
关键工序 |
工艺验证是验证一个确定的工艺,而非去研究一个工艺是否合适,因此在进行工艺前,工艺首先必须确定,必须在工艺验证前就确定了关键的质量指标和关键的工艺参数,而非是在验证后,再来确定这些指标。工艺验证不是工艺研发。
一般企业主要是在一定的方案下,连续生产三批商业化批量的原料药,质量和收率符合预定预期后,即认为工艺验证完成。
看了一些讨论,很多资料中写道:这种工艺验证仅仅是走形式,其表现为:1,对起始物料、中间体及终产品的关键质量属性没有把握住;2,对关键工艺参数识别不到位;3,验证过程各项指标的可接受标准制定依据不充分;4,对产生的合理偏差持否定态度,总想数据最完美。
单本人认为,什么QbD,风险评估毕竟还是一些大道理,能生产出来合格的产品本身来说就是好工艺。虽然工艺参数等确实需要认真研究,但完全没必要总是持自我否定的态度。比如碰到一个工艺就说:你这没有QbD啊。国内对QbD的研究文献,非常的少,大部分人讲的都是大道理,没有实际案例,很多都是空谈。比如ICH Q11提到的设计空间,一些控制参数不再是固定值,而是变量。
比如反应温度,30摄氏度的话需要反应6小时,40摄氏度的话需要5小时,50摄氏度的话需要3小时。60摄氏度的话,只需1小时就行了。而且不同温度,不同时间反应效果一样,超过时间则杂质量增多,无法去除。那么真实验证操作过程中,你敢把温度设定为30-60摄氏度,然后每小时取一次样吗?或者多批验证批次,每批的反应温度都不一样吗?
我觉得FDA的这套QbD理论,是先进的制药企业多年的经验总结,是一种制药文化精神的深入。我们才刚起步,目前需要的是更多的实例学习。而且就算要改掉药企以前工艺验证的弊端,也不能过急,需要慢慢进步。否则会起到拔苗助长的情况。
3)第三阶段:持续工艺验证阶段
该阶段,收集和评估所有生产中的信息与数据以评估工艺一直处在验证状态。这个阶段的产品已经开始上市销售,所以必须是满足GMP要求的。这个阶段数据汇总非常重要,如原辅料的质量情况、中间体质量情况、成品质量情况、每个生产步骤工艺、设备参数的变异情况、生产中的偏差、用户投诉、OOS等,从而分析出工艺的总体趋势。
中国GMP附录《确认与验证》第二十七条明确规定,在产品生命周期中,应当进行持续工艺确认,对商业化生产的产品质量进行监控和趋势分析,以确保工艺和产品质量始终处于受控状态。
简单来说,就是在以后的大生产中,也得时时刻刻保证着产品的质量稳定。不能有松懈。及时发现出现的新问题。
持续工艺确认与年度回顾和定期再验证的区别
|
与年度回顾的区别 |
持续工艺确认阶段并非是原来所有的回顾性验证或者年度回顾。它更加强调工艺验证的持续状态,也即持续的工艺验证并非只局限于在每年年底或者只是选择多少批的产品进行分析,而是时时刻刻的。 |
|
与定期再验证的区别 |
若企业尚未开展持续工艺确认,则应该定期开展工艺再验证。对己开展有效的持续工艺确认的企业,则不必要求工艺再验证。 |
总结
1、原料药工艺验证的问题,要研究的内容有很多很多。每个细节都关系着成败。
2、一些混批操作、中间操作留样量、下步投料量、三批验证连续操作的方式,都值得关注。希望有更多的权威部门发文,来明确这些操作的对与错。
3,中国是仿制药大国,更善于仿。很多问题,不是药企不认真去执行,而是没有思路或明确的指示。希望能有更多的,详细实际案例学习。例如ICH Q11刚颁布,美国的很多制药企业对里面的含义都不是很明朗,国内应该更难。如果只是单纯的对文件进行翻译,国内各家药企都摸着自己的石头过河,会很难达到预期效果。

来源:药事纵横


