您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2019-05-17 15:35
自从2011年FDA修订其工艺验证指南,到现在已有8个年头了,FDA已给企业充足的时间实施新指南,通过这一年多的FDA警告信显示,工艺验证的缺陷正集中爆发,原料药和成品药生产商应加强其持续工艺确认的方法。所以要认真的按三个阶段进行工艺验证,尤其强调持续的工艺验证。
工艺验证的要求也越来越严格,工艺验证要充分详尽,运用风险评估的方法确定关键工艺参数,制定详尽的验证主计划和持续工艺控制监测计划,以确保稳定的生产操作和持续的药品质量,运用有效统计工具识别出波动的来源,确保在产品生命周期内以持续方式对批间和批内变异进行监控,寻找改进的机会,同时在出现偏差时及时进行调查、风险评估及进行再验证。
一、各国法规监管的进程
2011年,当FDA修订其工艺验证指南时, 它引入了验证必须在制药产品的整个生命周期中持续进行的理念。
工艺设计,采用药品质量源于设计(QbD)的原则
工艺性能确认(PPQ)
持续工艺确认(CPV)
制药厂商一直在开发新的方法和解决方案,以支持这一工艺验证的新定义, FDA也一直在鼓励所有三个阶段的基于科学和风险的方法。
其他监管机构,包括国际协调委员会(ICH)在其Q8、Q9和Q10的问答(R4)行业指南中,也接受了工艺验证的生命周期方法。ICH将整个产品生命周期视为:开发、技术转移、生产和终止。
最佳实践需要以下方法:
采用实验设计(DOE)方法进行产品开发、取样和测试计划,可变输入与结果输出之间多种因素的相互作用来进行。
风险分析工具可用于筛选研究中的潜在变量,实现最小化实验来获得最大化的知识。DOE研究结果可以作为建立以后成品质量、设备参数以及中间体质量标准的依据。
工艺性能确认(PPQ)批次数量的确定。
使用合适的数据统计工具进行持续工艺确认(CPV)
工艺过程分析技术(PAT)的应用,实现设计、分析、和控制生产过程及时地测量。
欧洲药品管理局(EMA)2014年工艺验证指导原则、世界卫生组织的2015年附录7和国际药品监查合作计划(PIC/S)2015年附件15均反映了工艺验证应在整个工艺生命周期中持续进行的预期。
中国GMP附录《确认与验证》第二十七条明确规定,在产品生命周期中,应当进行持续工艺确认,对商业化生产的产品质量进行监控和趋势分析,以确保工艺和产品质量始终处于受控状态。
各监管机构都已认可这种全生命周期的工艺验证理念,对其要求也统一,并积极推动其发展,而制药厂商可以采取一种完整的生命周期方法来进行跨多个法规市场的工艺验证。
二、FDA警告信中关于工艺验证的缺陷分析
本文收集了从2017年至今FDA发布的警告信,通过分析FDA警告信(WLs),有助于制药厂商解决新的工艺验证需求。目标是了解哪些环节可能需要加强,以提高监管合规性并使风险最小化。
由于FDA工艺验证指导原则适用于原料药、制剂和生物制剂,警告信评估的见解可以提供充分的支持证据,以实现一种完整的工艺验证方法。
针对本文的目的,我们对2017年和2018年发布的85个FDA 警告信进行了分析,2017年61个,2018年33个。在原料药和成品制剂工厂中均发现了不足之处,但主要发生在成品制剂工厂中,如表I 所述。
表1 工艺验证缺陷一览表
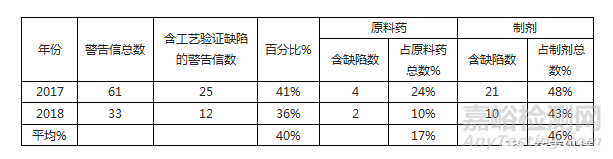
在2017年和2018年的FDA警告信中,工艺验证问题在成品制剂工厂比在API工厂更常见,在所分析的警告信中,40%包含与工艺验证相关的缺陷。自2017年以来,已发现31个成品制剂工厂在工艺验证方法方面不合规。
FDA警告信中主要提到的问题可分为两大类,具体如下:
1、未开展持续的工艺确认
未实施工艺性能确认研究,缺乏监测工艺控制的持续计划来确保稳定的生产操作和一致的药品质量。
应确定每种药品的工艺性能确认(PPQ)的时间表,描述你们持续监测批内和批间差异的方法,还特殊强调需要按照工艺验证(PV)指南的规定制定一个统计上合理的取样和测试计划。
一份以数据支撑的科学合理的工艺验证计划,恰当地识别出波动的来源,确保在产品生命周期内以持续方式对批间和批内变异的监管
2、前期的设计阶段或工艺验证阶段研究不充分,造成后续出现问题
没有表现出充分的工艺理解,其中包括未考虑关键操作的操作参数。
用于生产你们的XX药品的工艺未显示出其一致性和可靠性,你们药品的后续批次可能会在剂量、质量和纯度方面有重大变异。
缺乏工艺验证证据,因为你们未说明所有潜在的波动来源,这些必须受控以持续产出均一属性和质量的药品,且未证明工艺是可重复的。
总而言之,生产工艺的每个重大阶段都必须设计以确保原料输入、中间体和制剂成品符合其质量属性和标准。工艺验证评估一个工艺在其生命周期内的设计合理性和受控状态。工艺验证研究能确定是否已建立起初始的受控状态。在商业化销售之前要求有成功的工艺确认研究。之后,对工艺性能和产品质量要有持续的警惕监管,以确保你们在产品生命周期中维持稳定性的生产操作。
三、持续的工艺确认的方法
在预先确定好的期限,一般是一个月,确认该期限内该产品CPPs(关键工艺参数)、CQAs(产品关键质量属性)和CMAs(直接加入到工艺流中的关键物料质量属性)的受控情况,除符合合格标准外,可要对趋势进行分析。到期后根据对工艺受控情况的分析,增/减工艺控制策略,并按照新的策略开始下一周期的持续性工艺验证,以此循环。
注:CPPs、CQAs和CMAs之间的关系及识别的方法在这里不再阐述,已有很多文章进行了讲解。
四、持续工艺确认应该怎样做
由于行业对基于生命周期的工艺验证方法已经有了进步,现在一些公司将阶段1,2和3分为以下几个阶段:
阶段1A-产品设计开发,阶段1B-规模放大
阶段2A-设备、系统、公用设施、设施确认,阶段2B-工艺性能确认
阶段3A-对新工艺的强化确认程序,阶段3B-常规持续工艺确认。
在全球范围内实施工艺验证生命周期方法要求制药厂商在选择最有效的统计工具和阶段3策略时要特别注意。
一个好的知识管理策略是至关重要的,从工艺验证生命周期的第1阶段开始到第3阶段。科学地确定变异来源需要一个强大的基于QbD的产品开发计划,以及一个第3阶段允许持续地收集产品和工艺知识的CPV计划。
从过程漂移中学习,过程漂移或当工艺未能最大限度地执行时,触发识别变异来源,确保改进的控制,并允许持续改进。需要持续收集产品和工艺知识以持续改进,并改善类似产品的开发过程。
合适的统计工具应能非常直观将结果表现出来。能够识变异的来源,确定工艺改进的方向,无论使用何种统计分析工具,其目的都是对已有数据进行分析,以确认工艺处于受控状态或者识别工艺改进的机会。
五、总结
本文对警告信趋势进行了分析,为生产商提供了对监管机构目标的一些理解。在最近的检查中,由于违反了工艺验证指南,API和成品制剂生产场所一直被提到,而且国内的检查员也陆续对工艺验证提出持续工艺确认的要求。对缺陷的分析证实了需要一个定义明确的持续工艺确认计划,持续监测变异来源,在遇到质量问题之前,使组织能够进行持续改进。
同时我们不能为了验证而验证,要实际应用好,做好知识管理,用于实际的改进,世界各地的监管机构正在鼓励使用数据驱动的、以科学为基础的方法进行工艺验证。在某些情况下,检查员建议生产商寻求有资质的顾问的帮助,要求生产商投入时间和资源建立第3阶段CPV计划和支持团队。
现在更多的产商正在努力寻求数据统计的方法来进行第3阶段的工艺验证,解决方案的提供商正在提供集成的软件来帮助管理这样的程序。帮助所有组理解它们在促进持续改进中的作用。这种方法将有助于生产商防止过程失败,而且还可以防止人们对个别事件反应过度。它还可以确保它们捕捉到其他可能未被检测到的变异来源。
改进后必然要涉及到变更,做好后续的变更也至关重要,做好各种对比及质量研究,证明改进的可靠性。

来源:药事纵横


