您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2019-04-26 14:36
“包装”的通常作用,是保护产品、方便储运、促进销售,与产品本身相对独立,而最终灭菌医疗器械的包装,则是一类较为特殊的包装,直接影响到产品本身的质量安全,被管理机构定义为“医疗器械的一个附件或组件”,需要用一系列法规、标准进行规范。而对医疗器械企业而言,了解这些法规、标准就显得十分必要了。
最终灭菌医疗器械包装系统评价法规标准要求
国家总局制定并发布实施《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》(国家食品药品监督管理总局2014年第43号),对医疗器械注册申报资料进行了具体要求,尤其在“研究资料”要求提供“包装及包装完整性进行研究,在宣称的有效期内以及运输条件下,保持包装完整性的依据”。同时,国家也针对特定产品发布了指导原则指导企业准备注册申报资料,如《无源植入性医疗器械货架有效期注册申报资料指导原则(2017年修订版)》中对有效期验证内容进行了规定,要求验证“包装完整性,强度测试及模拟运输试验”。
《关于公布医疗器械生产质量管理规范附录无菌医疗器械的公告》(国家食品药品监督管理总局2015年第101号)和《医疗器械生产质量管理规范无菌医疗器械现场检查指导原则》(食药监械监[2015]218号),其中与无菌医疗器械包装系统相关的条款有2.6.1,2.10.1,2.11.1,2.12.1,2.13.1,*5.4.1,6.8.1,6.8.2,*7.20.2,上述9项条款对包装材料的仓储条件,包装工序的环境要求,初包装材料的生产环境洁净度要求,设计和开发输出中包装材料的技术要求,初包装材料选择和确认要求,初包装材料的初始污染菌和微粒污染可接受水平和控制要求和灭菌确认确定初包装的初始污染菌和微粒污染可接受水平都进行了明确要求。
最终灭菌医疗器械包装系统相关的包装标准已经形成系统。通用要求标准方面GB/T 19633(idt ISO 11607)系列标准,主要规定了最终灭菌医疗器械包装的材料、无菌屏障系统和包装系统的要求及包装过程(成形、密封和装配)的开发与确认要求。包装材料标准YY/T 0698(参考EN 868系列)系列标准,规定了最终灭菌医疗器械包装材料的具体要求和试验方法。测试方法标准YY/T 0681(参考ASTM标准)系列标准规定了无菌医疗器械包装的具体试验方法。包装标识标准YY/T 0461系列(idt ISO 15223系列)规定了医疗器械标签符号的要求。
无菌屏障系统评价
无菌屏障系统是“防止微生物进入并能使产品在使用地点无菌取用的最小包装”。其特有功能有:可对其进行灭菌、提供可接受的微生物屏障,可无菌取用。绝大多数无菌屏障系统在提供保护时将与产品直接接触。
结合无菌医疗器械生产质量管理规范及GB/T 19633.1要求,需要对无菌屏障系统的物理及化学性能、生物相容性和毒理学特性、微生物屏障、与成形及密封过程的适应性、与预期灭菌过程的适应性、与标签系统的适应性及灭菌前后贮存寿命进行评价。评价方式分为资料审核确认和材料测试,企业可根据供应商提供的证明资料,结合企业自身测试能力,进行风险分析后选择合适的评价方式。
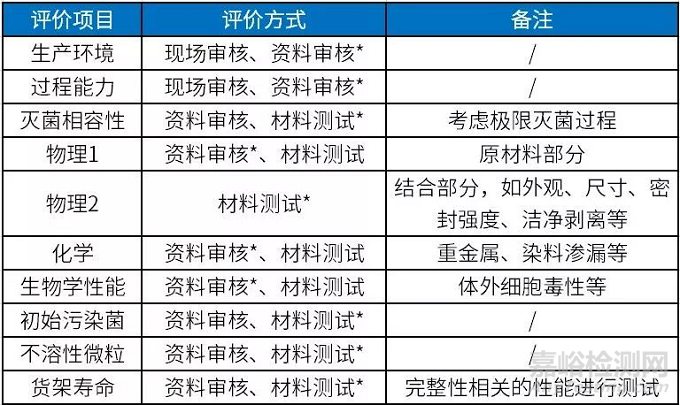
以某医疗器械为例,无菌屏障系统推荐评价项目可以包括表1中的评价项目。评价中使用的测试方法及接受标准可以参考YY/T 0698系列标准(最终灭菌医疗器械包装材料)和YY/T 0681系列标准(无菌医疗器械包装试验方法)。对于需要进行材料测试的评价项目,在进行样本量选择时,建议采用有统计学意义的样本量。
表1 无菌屏障系统推荐评价项目
*为推荐确认方式。
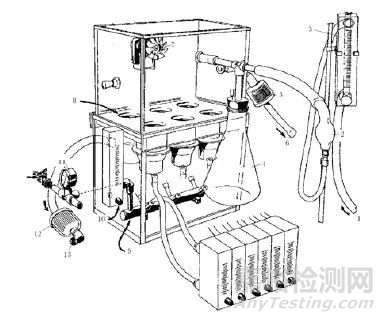
YY/T 0681标准中给出的微生物屏障分等实验装置示意图
最终灭菌医疗器械包装系统评价
最终灭菌医疗器械包装系统的目标是能进行灭菌、提供物理保护、保持使用前的无菌状态,并能无菌取用。最终灭菌医疗器械包装系统包括无菌屏障系统及保护性包装,在评价中应考虑器械预期的货架有效期、运输方式及存储要求。
结合注册审评研究资料及GB/T 19633.1要求,需要对最终灭菌医疗器械包装系统进行模拟运输性能试验及稳定性试验。模拟运输性能试验及稳定性试验的试验过程是对无菌医疗器械产品及包装的处理过程,试验的结果以无菌医疗器械产品及包装性能测试结果为判定依据。对于最终灭菌医疗器械需要评价的包装性能主要标签可识别性、无菌屏障系统完整性及强度测试。同样,测试项目样本量的选择时需要满足统计学意义。
模拟运输性能测试方法可以使用GB/T 4857系列(idt ISO4180)、ASTM D4169(YY/T 0681.15标准转换中)及ISTA 2/3系列标准任意一种标准中规定的方法,企业可以根据自己的情况自行选择。模拟运输性能试验设计时应考虑产品整个物流中的搬运方式、存储及运输情况,对于含药医疗器械等对储运环境敏感的产品,必要情况下先进行极限温度的验证(温度预处理)再进行模拟运输测试,极限温度验证的标准可以参考ISTA 2A、ASTM F2825(YY/T 0681.16标准转化中)及ISO 12417-1。待模拟运输测试包装系统的数量要求,首先要满足产品性能及包装性能测试样本量装载的包装系统数量,其次,若所有待测试产品只能装满一个包装系统的情况下,建议增加包装系统进行平行对比测试,多余需要的产品可以使用不影响测试性能的替代品进行填充装载。
稳定性试验应可以采用加速老化进行,加速老化的方法可以依据YY/T 0681.1(ASTM F1980)中规定的参数进行,但同时需要开展实时老化研究,在实时老化研究结果出具前,加速老化的研究结果可以被认为是标称有效期限的充分证据。在实时老化结果出具后,若实时老化研究结果不支持加速老化研究结果,建议首先分析异常出现的原因,在得到准确的证据后,进行风险分析,必要时,需要对已经放行的产品进行召回。
最终灭菌医疗器械包装系统是医疗器械的重要组成部分,是产品有效安全的重要保证。作为关键部件的无菌屏障系统的设计开发评价应纳入产品设计开发评价的范围,将法规标准要求融入产品的设计开发输入要求,并在输出时进行评价验证。在无菌屏障系统材料的选择上,使用已有验证的安全材料,并对封口过程进行确认,加工时进行参数控制,采取有效手段控制风险,才能切实保证最终灭菌医疗器械的安全和有效。

来源:王冠不掉


