您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2019-04-03 15:38
2018年底,口服制剂一致性评价期限已经到来,然而在200多个品种中,只有50多个品种目前通过,好在“大限”已经取消,下一步国家药监局如何行动,我们拭目以待。然而不管下一步策略如何,对于药品质量的提升的总体策略不会改变。对于当下的评价体系,不管是基于美国、中国的BE方案,还是基于日本的四条曲线,溶出都是重中之重,而其中药物制剂颗粒大小对于溶出曲线都会产生重要的影响。然而大家对粒度检测现在还是面临很多困惑,比如不同测试条件的结果有差异,但哪个条件更靠谱?为什么有的品种不同厂家结果差异巨大?谁家的粒度结果更靠近“真理”?今天小编就给您梳理一下。
1.1 为什么不同厂家结果会有差异,甚至有的差异巨大?
一般来说,两个方面的原因导致结果差异,一是光路结构和算法造成的。激光粒度仪是利用光学模型,通过米氏理论反演计算而得到的粒度分布,但每个厂家在开发仪器的时候,如何知道自己选用的模型是否正确?如何确定检测器位置以及光路是否合理准确?答案是利用粒径大小已知的球形标准粒子!因此每个厂家都是根据球形标准粒子来确定自己的模型和算法,从而一步步建立起来检测体系,一般球形的颗粒衍射光斑是一个个同心圆,而狭缝衍射光斑则是一组条纹。

球形粒子衍射光斑

狭缝衍射光斑
但我们真实的颗粒是啥样的呢?

乳糖
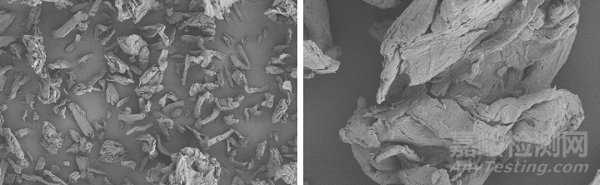
微晶纤维素
在这个背景下,各种颗粒形态各异,棒状的微晶纤维素,聚团的乳糖,不规则的各种API,球形的造粒等等,这也就意味着衍射光斑也会异常复杂,因而会导致计算结果产生各种“差异”。这也是为什么出现不同的粒度仪,大家测标样都非常帅,哪怕不一定都在不确定范围内,但肯定比较接近,可是一测实际的原辅料和制剂,那结果。。。。。。

API1 API2
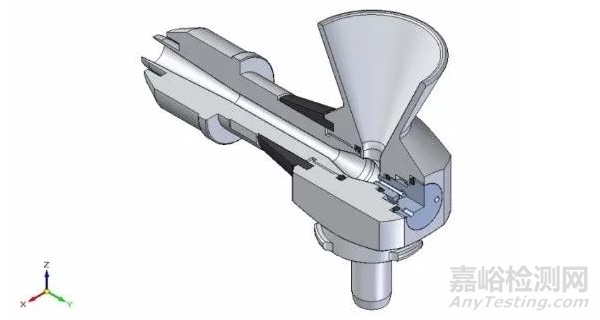
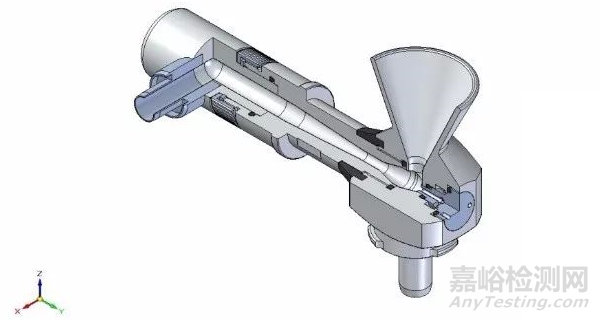
但有时候引起最大差异的还不在这,更大的差别可能来源于分散方式。一般药物颗粒分散有两种方式,即干法和湿法。每家的仪器都会配置分散附件,即将药物颗粒分散成单个的“粒子”然后通过检测器,然而由于设计、专利等方面的原因,导致各家在这一块能量输送有差异,比如湿法你家的循环管路、转速和超声输出可能跟我家就完全不同,甚至标称的一样输出功率,但里面结构不同,导致实际到颗粒的能量也有差异。如果说湿法还可以通过引进第三方(比如外置超声,分散处理完成后再加入到仪器中)来减少这种差异和风险,但干法就完全没有办法了,因为干法分散管路是跟仪器捆绑在一起的解决方案,是不可分割的。比如知名供应商A家其干法分散主要借助于不同的文丘里管路,早期的是通过软管连接,比如1号分散附件,后期的则通过直通管路连接,而供应商B则通过“龙卷风”加负压的方式,将颗粒吸入然后分散,而供应商C则直接将颗粒加压喷射于开放的空气中测试,这些处理方式特点各异,差异各异,正是因为这样,导致干法测试结果差异风险剧增。
A供应商型号1干法分散附件
A供应商型号2干法分散附件
A供应商型号3干法分散附件
B供应商干法分散附件

C供应商干法分散系统
有些朋友可能会立即心生疑惑,不管是哪种管路,只要都给我分散成一个个独立的“小颗粒”,那结果不应该都是接近的吗?的确,理想情况是这样,理论上也应该是这样,但偏偏实际上却不是这样,比如一个原辅料粉体,如果其粘附性很低,流动性很好,分布也不是很宽,那么这个结果确实应该接近,而且跟湿法也应该相差不大,但如果你的样品“比较粘”、“流动性不好”、“分布宽”,那你还认为你的颗粒出来结果还是一样?比如一款玻璃珠样品和一款阿托伐他汀钙API能一样吗?这也就是为什么在做药物颗粒粒度测试,一旦你选择干法解决方案的时候,你一定要清楚,这个样品可能是有管路依赖性的,压力依赖性的,不同仪器差异可能比较大的。
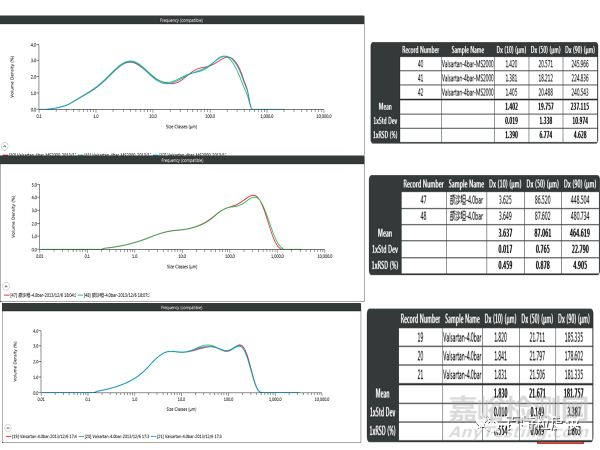
以上为一款结沙坦原料药干法测试结果,图中上中下分别为A供应商型号1干法附件、型号2干法分散附件以及型号3干法分散附件测试的结果,可以看到型号1分散附件测试结果D50在20微米左右,但型号2分散附件却有80微米,D90差异可能更大。但你可以下结论哪一种管路或者仪器测得更好吗?No,你没有办法证明,三个结果都做过压力滴定,都具有不错的精度。同时有的朋友认为可以通过图像来确认一下,但这种样品,颗粒从1微米不到,一直到几百微米,大家范围都是近似,但比例差异很大,你如何确定呢?粒度分布前面“鼓包”合理呢?无法确定,这就是现在颗粒干法检测面临的现状。
1.2 作为制剂或者原辅料厂家该怎么面对这种情况?
鉴于上面出现的情况,我们该怎么面对和处理这个事情呢?比如小编就碰到过这种情况,一款微粉化后的乳糖寄给3家不同供应商,结果回来一看,6微米、8微米、10微米,哇塞,等差数列呀!这个时候如果你纠结于6、8和10哪一个是真理的时候,可能就掉到坑里了,因为不管哪一个所谓巨头给你的测试结果,他都没法说明他的结果是“唯一的”、“正确的”、“接近真理”的。为啥?这乳糖不是标准品,形态又是各异的,图像一看也是差不多的,你能说明8微米比6微米准确?应该怎么办呢?怎么来考察呢?
接下来的问题就很重要,我们为什么要测这些颗粒?无外乎两个原因,第一个就是这个结果对我制剂工艺影响很大,不同大小的颗粒对我的混合、压片、溶出以至于最后的BE影响很大,所以我要控制它!!!第二个就是我是上下游供应商,我的原辅料要卖给制剂厂家,或者我要做入厂检测,因此我要比对数据,没这个数据我用不了。其实如果是这个过程,你需要的是什么呢?那就是这个测试结果精度是否够,其次能否反应我的工艺变化或者跟我的工艺产生相关性。或者就是一款原研的和仿制的放在一起,你能否客观给出这两个颗粒之间的差异,它俩是一样的吗?因此这个时候你更需要的工作,比如给你两款乳糖,这个工艺是有差异的,结果是有一定差别的,你能否在保证精度的情况下给我正确的对这两个样品进行区分。换句话说,我的一个原研的,一个仿制的其实是很接近的样品,你能否给我测出结果接近并且保证精度,第二就是我有差异的样品,这个差异你能不能给我区分和看到。而不是一个样品,寄给4个供应商,虽然可以做一些区分,但可能并不能从根本上解决你的问题。当然是不是我就只测试精度,客观性和准确性我就不用考虑啦?当然不是,那该如何做呢?那就是把方法学做细致,把验证工作做严谨做好!
1.3 如何来做方法学?如何做验证工作?
方法学很简单,其实就是考察可能影响你测试的所有影响因素,从中找到影响你测试粒度的关键质量因素(CQA),然后对这些因素进行风险评估和确认。确认了你的条件后,在进行一系列验证,包括精密度验证和交叉验证。比如通过图像技术、光阻法计数器技术等等,来考察你结果的合理性,比如你超声了,凭啥呀?你觉得干法将颗粒“打碎”了,凭啥呀?等等,这些证明并不是绝对的,但前提都是用数据说话。而不是我觉得谁家仪器很牛逼,谁的结果很好等等。当然告诉大家一些小的窍门,比如在送样考察环节,你可以给厂家不同的样品,其实标签是一样的,也可以给样品标签不同,但东西是一样的,可以给同样的样品两种工艺,差别比较细小的,也可以给两个差别比较大的,甚至混合一些样品,这些数据对于你判别合理性和以后你的工艺相关性都是大有帮助的,而不是单单给个标准球形颗粒,或者一个样品寄给4家的,这样就算结果回来,4个结果,然后你怎么办呢?
1.4 测试方式选择之争?到底干法还是湿法来做呢?
其实这也是客户很痛苦的,为啥呢?一款粉体我既可以用溶液或者悬液分散后直接测试,也可以用空气干法加压吹开测试。但往往这两种方式都是可以的,甚至药典也都是认的,但这两种方式该如何选择?怎样做风险更低?其实就目前市面上常出现的现状,比如干样干测学说,强调湿法可能会改变颗粒晶型,表面活性剂容易产生气泡,如果使用溶剂则产生废物,影响因素多难控制。而干样湿测学说则强调干法测试容易把颗粒“打碎”,细粉根本“吹不开”,控制因素偏少等。但小编觉得这都不客观的,举个例子,一款API,我明明是口服,药物释放明明在胃肠,都是在湿法的环境崩解和吸收,何来干法测试更加合理,干法没有破坏粒子?我又不是干粉吸入制剂DPI。但反过来,湿法测试学说,但有时候我很难找到一种好的溶液或者溶剂将颗粒悬浮怎么办?表活剂产生增溶或者气泡怎么办?多种原辅料,每种一个溶剂?溶解或者微溶怎么办?实际上很多时候这些学说都是供应商根据自家仪器的优势来选择,比如我家仪器干法强所以我选择干法解决方案,有的则湿法更强,所以推荐湿法解决方案。但我们究竟该怎么办呢?正确的方式是你要知道干湿法测试的特点,你要了解你家物料的特点。最后通过数据来确定你的测试方法,虽然我的数据也不是绝对的,选择也可能不同的,但一定要基于你的原辅料、你的制剂做出的判断,而且这种判断不是基于你的智慧和美丽,而是数据、数据、数据!当然实际操作中肯定要参考相关企业和以前的做法,这可以给你节省时间,但要牢记一点,隔壁王老五用的方法一定就得用,或者你就必须用,这个观点是站不住脚的。

最后,不管怎么样,面对越来越严的监管和形势的变化,希望我们国内的药企能够切实把制剂工作做好,药物质量越来越高,这才是能长久站稳脚跟的唯一方式。

来源:百特李雪冰


