1. 概述
普通的细菌毒素主要分成两种,一种是细菌外毒素(Exotoxin):它是一种有毒性的蛋白质,是细菌在繁殖过程中所分泌在细菌体外产生的有毒物质。产生外毒素的菌株,大多是革兰氏阳性菌,还有白喉杆菌、破伤风杆菌、肉毒杆菌、金黄色葡萄球菌以及少数革兰氏阴性菌。另一种是细菌内毒素(Endotoxin,脂多糖):为革兰氏阴性菌的细胞壁的产物,细菌在正常生活中时并不会自行产生,只有在细菌死亡自溶并附着在其它细胞上后,才显示其毒性。
细菌内毒素与人类生活息息相关,人类赖以生存的水源中同样含有细菌内毒素,其量约为1~100EU/ml。然而当细菌内毒素通过消化道进入人体时并不会产生影响,只有通过注射等方式进入血液时才会引起不同程度的危害。细菌内毒素少量进入血液后会被肝脏枯否细胞灭活,不会对机体造成损害,若大量进入血液后就会引起热原反应,即发热反应。因此,注射剂在产品研发上市过程中控制细菌内毒素很有必要。
2. 检查方法
根据中国药典2020版四部通则1143细菌内毒素检查法中描述,细菌内毒素检查包括两种方法,即凝胶法和光度测定法,后者包括浊度法和显色基质法。供试品检测时,可使用其中任何一种方法进行试验。当测定结果有争议时,除另有规定外,以凝胶限度试验结果为准。
目前,凝胶法检查法使用的较多,是一种限度检测或半定量的一种检验方法,光度测定法可定量检测细菌内毒素。
2.1 凝胶法
2.1.1. 实验原理
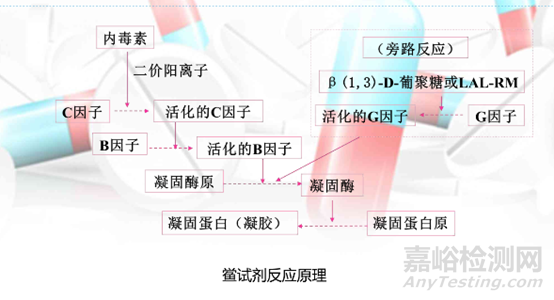
细菌内毒素的检查原理是通过观察鲎试剂与微量内毒素产生凝集反应的现象,来判断供试品中细菌内毒素的限量是否符合规定。
鲎试剂是一种无菌冷冻干燥品,由海洋生物鲎的血液变形细胞溶解物制成,其中含有能被微量细菌内毒素和真菌葡聚糖激活的凝固酶原,凝固蛋白原,能够检测样品中是否含有细菌内毒素和(1,3)-β-葡聚糖激,准确、快速地定性或定量。
反应原理如下:
2.1.2. 细菌内毒素限度的制定
一般情况下,是按照中国药典2020版四部通则1143细菌内毒素检查法进行计算:
3. 示例:XX注射液
3.1 限度制定依据
本品规格为300ml:API I 45g与API II 15g。参比制剂说明书中的常规用量用法为3~10min内滴注100ml,每1kg滴注7~20ml,每日API I最大剂量为200g,即本品每日最大剂量为1334ml。
根据上述公式,计算制剂的细菌内毒素限度L:L=K/M=5EU/(kg·h)/1334ml/(60 kg·h)=0.22EU/ml。
国家药品审评中心化药药物评价《细菌内毒素检查法研究中应注意的几个方面》中指出,大输液内毒素限度一般计为0.5EU/ml,所以拟定本品内毒素限度为0.5EU/ml。
3.2 物料控制
在注射剂中细菌内毒素贡献占比最大,需要根据产品的细菌内毒素来计算控制原辅料的细菌内毒素。故需要对本品原辅包的细菌内毒素制定合理的来源控制和过程控制:
3.2.1. API I
根据中国药典2020年版四部通则1143细菌内毒素检查法中公式,计算 API I的细菌内毒素限度L:L=K/M=5EU/(kg·h) / 200g/(60 kg·h)=1.5EU/g
式中:
L为供试品细菌内毒素限值,以EU/mg表示;
K为人每千克体重每小时最大可接受的内毒素剂量,本品为注射剂,K=5EU/(kg·h);
M为人用每千克体重每小时的最大供试品剂量,以mg/(kg·h)表示。人均体重按60kg计算,本品中API I临床每小时最大使用剂量为200g,故本品M=200g/(60kg·h)。
收紧细菌内毒素限度为≤0.5EU/g,并订入API I进厂内控质量标准。
3.2.2. API II
本品规格为300ml:API I 45g与API II 15g。按照参比制剂说明书用法用量,每日API I最大剂量为200g,即本品每日最大剂量为1334ml,故每日API II最大剂量为66.67g。根据中国药典2020年版四部通则1143细菌内毒素检查法中公式,计算API II的细菌内毒素限度L:L=K/M=5EU/(kg·h) / 66.67g/(60 kg·h) = 4.5EU/g
收紧细菌内毒素限度为≤0.23EU/g,并订入API II进厂内控质量标准。
3.2.3. 注射用水
参考ChP2020(注射用水细菌内毒素限度为≤0.25EU/ml),收紧限度为≤0.125EU/ml,并订入内控质量标准。
3.2.4. 内包材
直接接触产品内包材如西林瓶、胶塞、安瓿等,在生产使用时会要求除去细菌内毒素并进行验证,验证标准是内毒素能够降低3个log值。在产品细菌内毒素贡献占比较小。本品使用的内包材五层共挤输液用膜袋(含塑料输液容器接口和塑料输液容器聚丙烯组合盖)。参考FDA(≤0.5EU/ml),收紧包材组件的细菌内毒素限度为0.25EU/ml,并订入五层共挤输液用膜袋内控质量标准。
3.3 结论
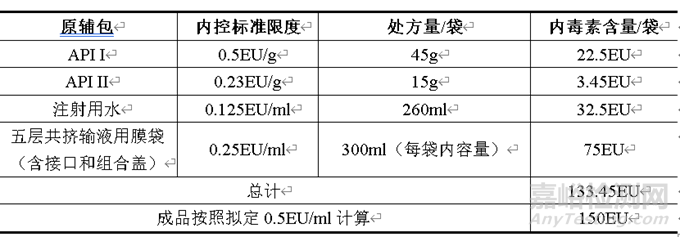
拟定本品的细菌内毒素限度为0.5EU/ml(即150EU/袋),原辅料、注射用水和内包材的细菌内毒素总量按照内控限度计算为133.45EU/袋,小于本品的拟定限度,计算结果具体见下表。从原辅料、注射用水和内包材方面可以有效控制本品的细菌内毒素,保证产品质量。
4. 实验过程
4.1 确定最大有效稀释倍数
确定供试品最大有效稀释倍数(MVD)。
MVD是指在试验中供试品溶液被允许达到稀释的最大倍数,在不超过此稀释倍数的浓度下进行内毒素限值的检测。(如果无限稀释供试品,内毒素可能会检测不出来。)
MVD=cL/λ
式中:
L为供试品的细菌内毒素限值;
c为供试品溶液的浓度,当L以EU/ml表示时,则c等于1.0ml/ml,当L以EU/mg或EU/U表示时,c的单位需为mg/ml或U/ml。
λ为鲎试剂的标示灵敏度(EU/ml)【凝胶法】,或是标准曲线最低的内毒素浓度【光度测定法】。
4.2 鲎试剂的灵敏度复核试验
鲎试剂的灵敏度(EU/ml)为在检查法规定的条件下,使鲎试剂产生凝集的内毒素的最低浓度。
如果使用新批次的鲎试剂,或者试验条件发生了改变时,包括任何可能影响检验结果的改变,都要进行鲎试剂灵敏度复核试验。复核的目的是为了确认鲎试剂的灵敏度,同时也考察了检验人员操作方法,并确认了试验条件是否符合规定。
4.3 干扰试验
干扰试验是为产品使用细菌内毒素检查法提供依据,确定供试品在确定浓度下对内毒素和鲎试剂的反应都不存在干扰作用。在开发细菌内毒素的检查方法时,试验前须进行干扰试验。当试验条件发生了改变时,包括鲎试剂、供试品的配方、生产工艺改变或试验环境中发生了任何变化时,须进行干扰试验。
4.4 供试品检查
使用已经过验证的方法对制剂品种进行检验。
5. 小结
在注射剂的生产过程中,可能会从原辅料、生产过程、人员、设备以及环境等各个方面引入微生物及细菌内毒素。为了保证药物使用的安全性,各国法规标准对于注射剂的细菌内毒素水平都有严格的规定,若没有标准,也需要在研发过程中制定相应的内控标准。也同时要求生产企业在注射剂研发过程中采取合理的策略,严格控制注射剂中的细菌内毒素含量。