您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-10-15 08:55
为解决临床急需问题,促进药物可及性,中美欧采取了不同的加快药品上市的注册程序。
中国药品加快上市注册程序
《药品管理法》《疫苗管理法》及国务院文件中列明的临床急需的短缺药、儿童用药、罕见病用药、重大传染性疾病用药、疾病防控急需疫苗和创新疫苗等,均被明确纳入加快上市注册范围。结合我国行业发展和公众用药实际,参考国际经验,新修订的《药品管理法》以及《药品注册管理办法》(以下简称新《办法》)提出了四种药品加快上市注册程序,分别是突破性治疗药物程序、附条件批准程序、优先审评审批程序以及特别审批程序,并对适用范围、工作程序、审评时限进行明确规定。目前,突破性治疗药物程序、附条件批准程序、优先审评审批程序均于2020年7月8日开始试行。其中临床急需药品附条件批准上市技术指导原则于2019年11月8日再次征求意见。特别审批程序仍参照2005年11月18日颁布的《国家食品药品监督管理局药品特别审批程序》中的相关规定执行。
四种加快审评程序具体要求见表1~表4。

图1:中国药品加快程序申请时间决策图
美国药品加快上市注册程序
针对未满足的临床需求,美国FDA在药品审评过程中,有多种加快审评审批路径,包括快速通道(Fast Track)、突破性疗法(Breakthrough Therapy)、加速审批(Accelerated Approval)和优先审评(Priority Review),在《FDA行业指南:严重疾病加快程序–药物和生物制剂(2014)》(FDA Guidance for Industry: Expedited Programs for Serious Conditions – Drugs and Biologics (2014))中有详细介绍。加快审评通道覆盖了从临床早期到上市批准的各个阶段,极大促进并加快了创新药物的研发和上市。

图2:美国药品加快程序申请时间决策图
欧盟药品加快上市注册程序
自2006年开始,欧盟先后建立了附条件上市许可(Conditional Marketing Authorization, 2016年修订)、优先审评(Accelerated Assessment, 2004年)、适应性审评(Adaptive Pathway, 2014年启动)、重点药物审批(Priority Medicine, PRIME,2016年启动)加快注册程序。

图3:欧盟药品加快程序申请时间决策图
中美欧药品加快上市注册程序对比分析

表1 突破性治疗相关程序对比
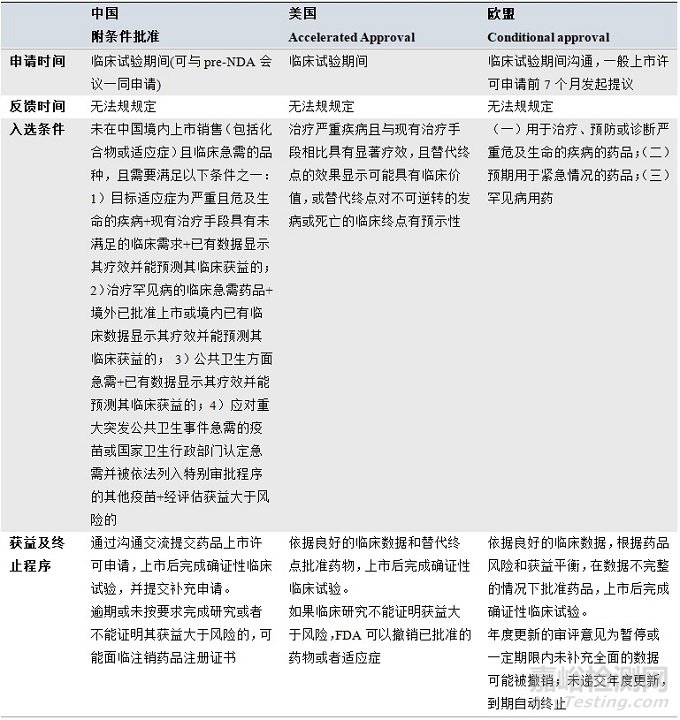
表2 附条件批准相关程序对比

表3 优先审评审批相关程序对比
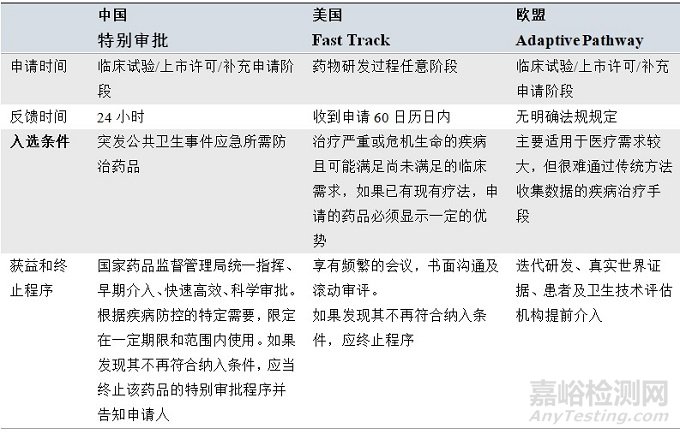
表4 几种特有审评程序对比
我国的药品加快上市程序参考了欧美相关规定,但又具有中国特色。我国与美国的突破性疗法的适用范围都比较广(新分子和新适应证),而欧盟的PRIME程序只适用于新药(新分子),欧盟每月都会公布纳入PRIME的药物。美国的突破性疗法认定不公开,而我国在新《办法》中加入了对申请突破性治疗药物的公示程序。截至2020年9月30日,我国已有5个品种正式纳入突破性治疗药物程序,有另外2个品种处于公示中;共计7个品种,其中有6个品种为已进入2期的抗肿瘤产品,其中2个境外生产品种也已获得FDA的突破性治疗药物认定。
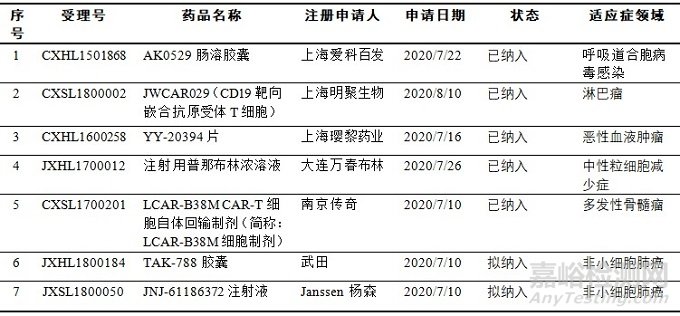
表5:我国纳入突破性治疗药物程序的产品列表
我国的附条件批准与美国的加速审评、欧盟的附条件上市许可相似,获得阶段性良好数据后批准药品上市,上市后完成确证性试验。美国附条件批准主要是基于替代终点的批准;欧盟根据风险和获益作出批准决定,即产品上市对公共卫生带来的益处大于缺少详细数据带来的风险;我国的附条件批准规定申请人必须履行特定条件的情况下基于替代终点、中期分析、早期临床试验数据或境外临床试验数据而批准上市,上市后进行确证性临床试验。虽然附条件批准程序2020年7月才正式试行,但此前,国家药监局和CDE已有按附(有)条件批准的相关产品,如2018年4月有条件批准用于预防宫颈癌的九价人乳头状瘤病毒疫苗;2018年12月有条件批准首个国产PD-1治疗既往标准治疗失败后的局部进展或转移性黑色素瘤的特瑞普利单抗;2019年11月有条件批准了治疗阿尔茨海默症新药甘露特钠胶囊;2019年12月附条件批准了卵巢癌治疗药物甲苯磺酸尼拉帕利胶囊;2020年6月有条件批准了泽布替尼胶囊等。在更早以前,国家药监部门和CDE也使用附(有)条件批准的理念批准了部分临床急需的药品。
我国的优先审评审批与美国的优先审评和欧盟的加速审评相似,在申报上市时提出申请,缩短审评时限。在适用范围上,美国适用面较广,涵盖了治疗和技术方面的创新和改良。而我国作了更具体的规定,主要包括临床急需的短缺药、儿童用药、罕见病用药、重大传染病用药等,更强调有突出临床价值与临床急需药品,且仅限于上市申请,从而进一步提高审评审批效率。CDE网站显示,截至2020年9月30日,共有1177个品种获准进入优先审评审批程序。此外,国家药监局于2018年和2019年分别发布了两批《临床急需境外新药名单》,建立专门审评通道,大幅加快境外已上市临床急需新药进入我国。
中美欧特有审评程序
我国的特别审批程序经常会和美国的紧急使用授权(Emergency Use Authorizations ,EUA)混淆,其实二者有很大差别。特别审批程序是指存在发生突发公共卫生事件的威胁时,为使突发公共卫生事件应急所需防治药品尽快获得批准,遵循统一指挥、早期介入、快速高效、科学审批的原则,是上市许可中一条常规的通道,药品被批准后长期有效。对于2019年末暴发的新冠肺炎疫情,我国按照上述四原则,快速高效批准了多种检测试剂盒,对于潜在治疗药物、治疗新冠抗体或预防用疫苗等药物的审评工作以小时计,甚至从注册递交到启动临床试验仅用短短几天的时间。美国的紧急使用授权(Emergency Use Authorizations ,EUA)是美国一条非常规的审批通道,属于阶段性的上市途径,美国EUA允许FDA通过促进公共卫生紧急情况下所需MCMs (Medical countermeasures)的可用性,帮助FDA加强针对CBRN (chemical, biological, radiological and nuclear) 威胁的国家公共卫生保护,国家度过紧急状态后药品的上市许可可能面临被撤销。我国《疫苗管理法》第二十条也有相应的规定,“出现特别重大突发公共卫生事件或者其他严重威胁公众健康的紧急事件,国务院卫生健康主管部门根据传染病预防、控制需要提出紧急使用疫苗的建议,经国务院药品监督管理部门组织论证同意后可以在一定范围和期限内紧急使用”,但目前还没有正式落地政策出台。
美国的快速通道,是目前各国所有程序中申请时间最早、覆盖范围最广的程序,最早可通过非临床数据,在临床试验前申请,但实际操作中逐步被突破性疗法取代。
欧盟的适用性路径是努力促进患者及时获得新药的重要举措,其主要适用于医疗需求较大,但很难通过传统方式收集数据的疾病治疗手段。
综上,新《办法》明确了四条药品加快上市通道,其在适用范围和加快程序等方面与欧美加快通道有很多相似之处,同时也存在一定差异。随着新《办法》及相关加快通道指导原则的正式实施,预计具有临床价值和临床优势的创新药物在我国的研发及上市将大幅加快,也将有更多的境外新药在中国与全球同步研发上市。

来源:中国医药报


