您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-05-07 17:05
为确保药品质量和防止交叉污染,生产设备的清洁方法应当经过验证。随着 FDA 对清洁验证更加重视,验证中可能存在各种问题和困难。文章将基于制药企业的视角,讨论清洁验证实施过程中需重视的问题,以及需要采取的应对策略。
1、清洁验证概述
清洁验证目的是证明生产设备经过清洁,将化学残留、微生物残留清洁至可接受限度,避免产生交叉污染。清洁验证全过程考虑其生命周期[1],药品生产过程应有一个经验证且受控的设备清洁程序,制定合理的接受标准,对清洁效果持续监测,确保方法持续有效。
1.1清洁验证应具备的条件
1.1.1 清洁方法的开发与建立
清洁方法开发与建立的内容包括选择合适的清洗剂、清洁方法的设计和可操作性等。
应用风险评估[2]来确定验证的水平及清洁规程的要求。风险评估至少包括以下内容:活性物质,辅料和中间体的溶解度;剂量水平/毒性/药效;设备的设计和构造,包括产品直接/间接接触的设备表面;专用/非专用设备;关键的设备部件;设备表面材料的吸附性;清洁的难点;清洁周期,特别是专用设备,用来预防分解物质残留的累积;微生物、内毒素的风险。
1.1.2 可接收标准和分析方法的验证
可接收标准:生产设备存在化学物质 (活性药物成分、辅料、洗涤剂、消毒剂)、微生物、内毒素污染。这些污染物限度首先应目视设备清洁无可见异物。
化学残留可接收标准:采取以下三种计算方式进行 MACO(允许最大残留) 的计算,再依据 MACO 计算可接受限度。第一种:据产品的 ADE(可接受日暴露值) 或 PDE(允许日接触量)计算;第二种:根据 TDD(日治疗剂量) 计算;第三种:根据一般限度标准 10~1 000 mg/L 之间。对于毒性、过敏性、致畸性的产品限度应当根据风险评估制定,通常规定为 10 mg/L。选择这三种残留最低限度作为清洁验证可接受标准。
微生物可接收标准:接触和擦拭法可根据环境监测规程中相应的表面微生物要求。淋洗法:根据最后所用淋洗水的微生物要求。
内毒素可接收标准:有内毒素要求的产品设备清洁后一般需考虑内毒素残留,如果已清洁设备后续还有去除内毒素的方法,则不需要考虑内毒素残留。选择产品内毒素标准作为可接受标准。
分析方法验证:不同设备的材质应经过回收率验证,样品的回收率和重现性必须通过实验进行测试,按照不同的材料类型和表面处理情况来进行取样以表明设备表面的污染物具有重现性,擦拭取样的回收率必须大于或等于 50%。分析方法应考虑专属性或非专属性来显示清洁方法的有效性。
1.1.3 强化人员培训
关键区域手动清洁的设备,应借助高压喷头的专用工具进行清洁,这些特定设备有不同的清洁程序。人是最不确定因素,手动清洁因操作人员不同而有差异性,为了证明清洁方法的可靠性,清洁验证时应包括不同的员工,且由经过培训的人员来执行。
2、制药企业需要重视的问题与应对
2.1忽视法律法规对设备清洁的要求
设备清洁是一个非常重要的内容,有很多公司都忽视了法律法规中对设备清洁的要求,曾经收到过“警告信”。图 1 中列出了从 2002 年到 2006 年 5 年间发生的 483 缺陷的统计情况。值得注意的是,将近 95% 的“警告信”都是因企业没有制订出有效防止潜在交叉污染的设备清洁程序而产生的。没有定期对设备和器具进行清洁与维护,有可能造成交叉污染,并对药品的安全性、鉴别特性、浓度/效价、质量和纯度造成影响,违反联邦法规 21 第 211.67a。
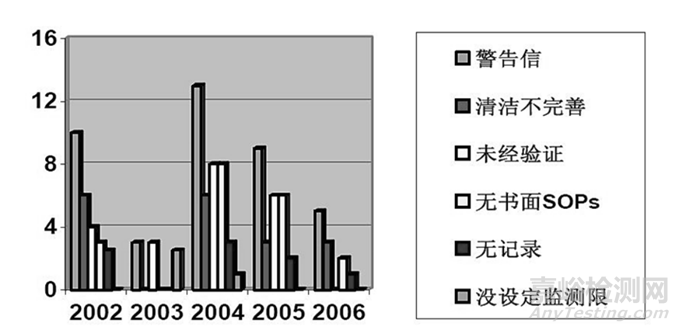
图1 2002年到2006年483缺陷的统计
生产中所用设备 (包括器具) 的清洁维护没有建立适当的规程。违反联邦法规 21 第 211.67b。
没有保存药品生产中所用设备的维护、清洁、消毒和检查的记录,无设备清洁、维护、使用的记录,包括日期及使用时间。违反联邦法规 21 第 211.67c。
清洁验证中产品接触的表面材质如不锈钢、搪玻璃、硅胶、有机玻璃、聚四氟乙烯验证没有涵盖全面,擦拭面积、擦拭方法没有与验证一致等。
2.2化学残留限度很低及应对
当计算所得的化学残留限度低于标准限度时,有以下方法应对:
(1) 使用灵敏度高的检验方法。分析方法多种多样,有特定的分析方法 ( 如:HPLC、UPLC 等 )、非特定的分析方法 (如:TOC、UV、电导率或 pH等),多功能车间精制步骤一般选择特定的分析方法。检测清洁剂可以采用选择性的方法或者非选择性的方法,例如 TOC。
(2) 提高清洁验证时的取样样品浓度或其他办法,更改取样参数。例如擦拭取样中,通过增大取样面积或者减少拭子萃取的溶剂量。
(3) 提高清洁验证时的取样样品浓度或其他办法。
(4) 减少淋洗量。
(5) 增大后续产品的最小批量会提高限度。
(6) 限制生产顺序,如果已清洁的产品残留限度较低,则下一生产产品不能为限度更低的产品。
(7) 利用“ICH M7 基因毒性杂质限度确定”提高 TTC。
2.3取样不具代表性及应对
清洁验证中常见的取样仅为淋洗液或仅为擦拭法。两种方法各有优缺点。淋洗的优点是它能测量整个设备的表面区域。然而,选择的溶剂必须确保污染物的高回收率。但弱点是污染物可能不会溶解或可能遇到稀释问题,因此不能确保检测的准确性。
擦拭法的优点是提供了一个表面的直接处理方法,但它不适用在大型设备的完整表面取样,所以必须选取有代表性的取样部位。这个方法也有技术局限性,不适用于那些无法取样的设备。擦拭适用于无法进行淋洗取样或回流溶剂时的取样,如:打粉和筛网、包装袋、托盘干燥器。
擦拭法应对取样部位最难清洁[3] 的部位进行评估,通过验证证明哪个部位最难清洗,取样点选取应坚持以下原则:最有代表性的部位、关键部位、各类最差点 (难清洁的部位)、与产品直接接触的表面、气流型导致的最差点、活动频繁的地方、靠近产品的地方、结构材料不同的部位、取样点的方便性和重现性。
2.4取样污染及应对
取样时先取空白棉签,空白棉签先用相应的溶剂润洗,与样品隔离;同一个取样点应该先取微生物,再取化学残留,取微生物时不能在同一点取化学残留的样品,但必须在同等水平的点上取样,微生物取样后应清洁该取样点;取样容器如用于微生物残留测定,应选用已经过灭菌的取样瓶;如用于化学残留,应确保取样容器的清洁;对于所有清洁验证的设备如反应釜,过滤器和干燥器等应在淋洗之后取擦拭样;如果仅擦拭样或仅淋洗取样,需要给出充分的理由,证明取样具有有效性和代表性;取样擦拭方法应与验证方法一致。
2.5手动清洁的差异及应对
对手动清洁的设备建议每年进行一次随机检查。由车间启动随机检查,由质管部批准。在检查过程中,最好检查被认为是“难以清洁”设备的部分,必要时检测化学残留或微生物残留,每年年初初次验证时和有新员工参与时,可用产品进行模拟污染、模拟清洗。
3、实施成功的清洁验证总策略
在设备方面,确认设备不存在清洗死角,具备合适的喷淋、干燥功能,CIP 程序等。
在确认清洁方法方面,确认清洁方法合理,具有重复性、可操作性,若设备共线,应确认最难清洗产品、最差条件 (如:溶解性、毒性),确认下一产品剂量最大、批量最小。
在文件准备方面,查阅清洁验证 SOP,计算面积、统计材质、计算限度、分析方法开发和分析方法验证。
4、结语
清洁验证既是保证患者用药安全的一个重要要素,通常也是 FDA 检查中的关键项。本文阐述了清洁验证实施过程中遇到的常见问题,制药企业应结合自身情况采取相应策略,不断完善和改进清洁验证工作。
参考文献
[1] 翟铁伟. 药品生产中清洁验证的生命周期探讨[J]. 中国医药工业杂志,2019, 50(11): 1341-1347.
[2] 姜斌. 质量风险管理在非无菌原料药清洁验证评估中的应用[J]. 中国药师,2017, 20(12): 2281-2285.
[3] 高歌,张会云,尤春玲,等. 制药企业共线生产产品清洁验证[J]. 质量探索,2016, 138(4): 46-47.
本文作者丁婷婷,浙江医药股份有限公司新昌制药厂,来源于化工管理,仅供交流学习。

来源:Internet


