2023年12月,FDA发布了《clinical pharmacology considerations for peptide drug products-guidance for industry》的草案,恰逢现在全球GLP-1R、GCGR等代谢类靶点火热,多肽类药物研发骤增。结合该指导原则关键要点及相关案例,略作分享、讨论。
多肽既可以从动物组织分离,又可以化学合成,还可以通过重组表达获得。多肽药物研发趋势正在往ADME属性改善的方向发展,常见做法包括增加口服生物利用度、延长半衰期、降低整体疏水性、增加构象灵活性以增强目标靶点的选择性等。可以通过环化、伪肽键、非天然氨基酸和多肽修饰如PEG化等路径实现以上目的。也正是基于以上改造策略,使得多肽药物的PK、PD等行为有别于传统小分子和生物药。
FDA在指南中先来了点开胃菜,再进入的正题。共三道开胃菜:1)生物分析方法需要全面验证,具体可参照ICH M10;2)如果多肽基于临床前数据,代谢和排泄路径比较清楚,临床药理部分可以不开展放射性标记的物料平衡研究,具体可以参考《Clinical Pharmacology Considerations for Human Radiolabeled Mass Balance Studies》;3)分子量小于69kDa的多肽或蛋白药物,需要开展肾损伤患者的PK。
正题部分,FDA主要分为4个主题:免疫原性、肝损伤影响、药物-药物相互作用、QT间期延长考察,逐一细表。
免疫原性
首先,评估免疫原性风险。可以参考《Immunogenicity Assessment for Therapeutic Protein Products》,从产品自身因素(如分子量、结构)、工艺相关因素(如宿主细胞蛋白)、患者因素(如疾病状态)、临床方案设计(如给药途径、联合用药)等角度综合考量、评估免疫原性风险。一般来讲,氨基酸数量小于8个的多肽药物,并不认为具有免疫原性。
其次,对药物的免疫原性进行检测。检测方法可以参照《Immunogenicity Assessment for Therapeutic Protein Products》和《Immunogenicity Testing of Therapeutic Protein Products — Developing and Validating Assays for Anti-Drug Antibody Detection》,这点与常规蛋白类药物没有太大区别,都是按照多级检测原则,筛选、确认、滴度和中和活性测定。FDA特别提了两点,一是如果多肽药物有多个结构域,比如Tirzepatide,可以考虑开发多个方法检测针对不同结构域的免疫反应。第二,如果多肽药物与内源性物质具有同源性,建议对抗药抗体与内源性物质的交叉反应进行考察。同样以Tirzepatide为例,其Ⅲ期临床研究结果显示,66% ADA阳性患者出现与天然GIP的交叉结合,27%出现与天然GLP-1的交叉结合。
最后,评估ADA结果对PK、PD、有效性和安全性的影响。主要从两个角度着手,一是个体间,即ADA阳性和阴性个体间的对比。二是个体内,即ADA阳性前和阳性后之间的对比。除了对比ADA阳性、阴性的影响,还需要进一步评估抗体滴度、中和抗体对PK、PD、有效性和安全性的影响。
肝损伤的影响
多肽通常经由内肽酶和外肽酶代谢为氨基酸。由于蛋白酶和肽酶在全身无处不在,许多肽的蛋白水解降解是快速的,并且不仅限于药物消除相关的器官,如肝脏。因此,肝脏在肽类药物清除中通常并不发挥主要作用。但FDA给出一些特殊场景,建议出现以下情况时考虑开展药物在肝损伤患者中的相关研究:
1)非临床研究结果显示,多肽类药物经肝药酶代谢比例超过20%;
2)对多肽药物的某些修饰,比如环化,会增加肝药酶代谢的敏感性;
3)临床前研究(如胆管插管动物实验)显示,>20%原型药或活性代谢产物通过胆汁排泄。这种情况,即使药物不经肝药酶代谢,肝脏的损伤也会影响药物暴露情况。
4)偶联脂质基团(如脂肪酸或胆固醇)多肽药物可以与体内白蛋白或其它脂质成分结合,延长作用时间。但如果患者有慢性肝脏疾病,会导致血清白蛋白水平下降,药物清除有加快风险;
5)如果肽类药物临床适应症是肝脏疾病或者具备靶向肝脏的特点(如分子中含有GalNAc),那么肝损伤或许不影响PK,但可能对PD产生影响;
药物-药物相互作用
肽类药物作为CYP肝药酶和转运体的底物
多肽类药物主要经内肽酶、氨肽酶或羧肽酶等蛋白酶水解,不被CYP450肝药酶代谢。因此,肝药酶的抑制剂和诱导剂理论上不会影响多肽类药物PK。同样的,肽类药物的PK一般也不受各种转运体的影响。
但事无绝对,还是有些情况需要考虑肝药酶和转运体的影响。如果肝脏和/或胆汁排泄≥20%总消除量,或者药物靶器官为肝脏时,建议开展体外CYP450酶、转运体相关的研究。如果肾脏排泄≥25%总消除量,或者药物有肾毒性时,建议开展体外肾脏转运体如OAT1、OAT3、OCT2、MATE11等相关的研究。
肽类药物作为CYP肝药酶和转运体的抑制剂或诱导剂
一般来讲,肽类药物对CYP肝药酶和转运体没有调节作用。但是,如果多肽药物经过修饰,比如环化,则有可能对CYP肝药酶和转运体产生影响。有些影响是间接的,比如生长抑素类似物(如lanreotide、octreotide)可以调节CYP酶的表达,从而降低一起用药的其它药物的清除。具体可以参考《In Vitro Drug Interaction Studies-Cytochrome Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry》、《Clinical Drug Interaction Studies-Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry》。
还有些多肽药物因其作用机制特别,也会改变共同用药的药物PK。比如GLP-1受体激动剂(如艾塞那肽、利拉鲁肽、司美格鲁肽等)具备延缓胃排空的药理作用,如果联用口服药物,则会对这类药物的PK产生影响。
QT间期延长风险
肽类药物对离子通道产生影响的可能性很低,除非在临床或非临床研究中观察到心律失常风险,一般不需要开展详细的QT研究。但是,需要按照ICH E14、ICH S7B等指导原则要求将评估的QTc间期延长的风险、计划开展的QTc监测计划提交给监管机构。
案例解析-司美格鲁肽
1)指南:如果多肽基于临床前数据,代谢和排泄路径比较清楚,临床药理部分可以不开展放射性标记的物料平衡研究。
司美格鲁肽临床前其实是开展了很完整且深入的代谢和排泄研究。但临床药理研究中,诺和诺德依然在7例健康男性中开展了3H标记的司美格鲁肽皮下给药代谢和排泄研究(研究代号:NN9535-3789)。
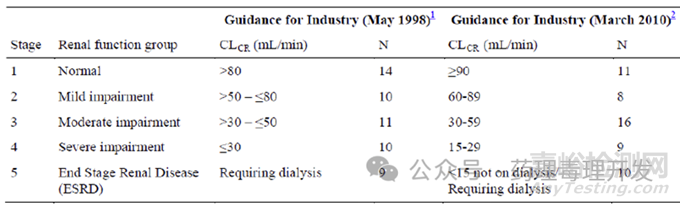
2)指南:分子量小于69kDa的多肽或蛋白药物,需要开展肾损伤患者的PK
司美格鲁肽的分子量4.1kDa,符合开展肾损伤患者PK的要求。因此,如下表所示,根据肌酐清除率,将患者分为肾功能正常、轻度、中度、重度和终末期肾损伤级别,每组9-16例患者,考察了肾功能损伤程度对司美格鲁肽PK的影响。试验分两阶段开展,先开展正常肾功能患者、终末期肾损伤患者的研究,如果结果显示对PK无影响,则不需要继续开展第2阶段轻度、中度肾损伤患者的研究。反之,则需要开展第2阶段的试验。当然,结果显示肾损伤对司美格鲁肽的PK没有影响,即有肾损伤的2型糖尿病患者不需要调整剂量。
3)指南:偶联脂肪酸链的多肽考虑开展肝损伤对PK的影响
司美格鲁肽偶联脂质基团脂肪酸,可以与体内白蛋白结合,延长作用时间。如果患者有慢性肝脏疾病,会导致血清白蛋白水平下降,药物清除有加快风险。因此,司美格鲁肽开展了在肝功能正常、轻度、中度和重度肝损伤患者的PK研究。结果显示,肝损伤对司美格鲁肽的PK没有影响,即有肝损伤的2型糖尿病患者不需要调整剂量。
4)指南:免疫原性研究
诺和诺德开发了检测抗司美格鲁肽抗体的方法。32例(1%)给予司美格鲁肽的患者ADA阳性,其中19例(0.6%)患者形成的抗体与内源性GLP-1有交叉结合。
5)指南:药物-药物相互作用:肝药酶和转运体的底物、抑制、诱导及共同用药的影响
肝药酶底物验证:开展了两项体外肝细胞研究。1)3H-Tyr-semaglutide在Wistar大鼠、食蟹猴和人肝细胞单层细胞孵育4、24h,发现司美格鲁肽非常稳定,人和猴中100%以原型存在,大鼠中>99%是原型药物。2)3H-Oct-semaglutide在SD大鼠、食蟹猴和人冻存肝细胞孵育4h,分别有83.7%、96%、94.5%以原型药物存在。因此,司美格鲁肽不是肝药酶的底物,具备肝药酶代谢稳定性。
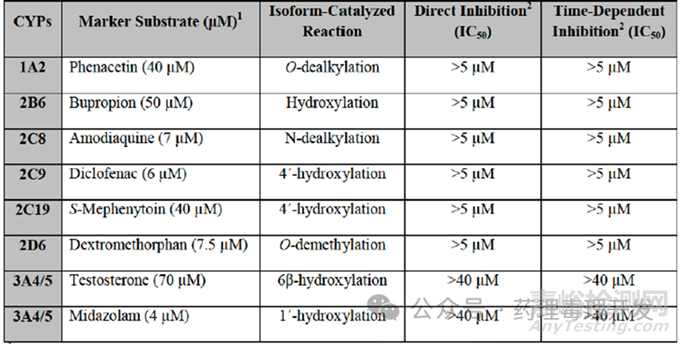
肝药酶抑制风险:体外采用人冻存肝细胞研究了司美格鲁肽对CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6、CYP3A4/5代谢各自底物的抑制能力。结果如下表所示,司美格鲁肽对上述各酶的活性抑制IC50均>5μM,风险很低。
肝药酶诱导风险:体外采用人冻存肝细胞研究了司美格鲁肽对CYP1A2、CYP2B6、CYP3A4/5代谢各自底物的诱导能力。从两个层面进行了研究,一是检测底物特定marker,二是检测酶的mRNA水平。结果显示,司美格鲁肽对3种酶的诱导作用在>0.5倍和<2倍之间,风险比较低。
转运体抑制:体外研究了司美格鲁肽对ABC转运体P-gp、BCRP,SLC转运体OATP1B1、OATP1B3、OAT1、OAT3、OCT2的抑制作用。P-gp采用Caco-2细胞,BCRP采用MDCKII-BCRP、SLC转运体采用HEK293细胞进行验证,设置合适底物和阳性对照。结果显示,司美格鲁肽对P-gp、BCRP、OCT2、OAT1和OAT3没有抑制作用。不过,对SLC转运体中的OATP1B1和OATP1B3的转运底物能力是有一定抑制作用的,但IC50分别为3.5和2.95 μM,超过临床稳态Cmax浓度33 nM大概100倍,因此,预期临床不会对这两个转运体产生抑制作用。
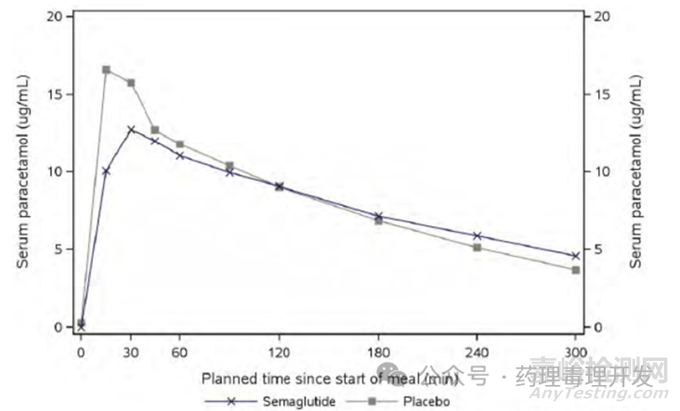
联合用药相互作用:GLP-1受体激动剂的作用机制之一就是减缓胃排空。因此,与口服药物联用就可能影响其PK行为。诺和诺德研究了司美格鲁肽对扑热息痛PK的影响,结果如下图所示,司美格鲁肽降低了23%的扑热息痛Cmax,总的AUC影响不大。
诺和诺德还研究了司美格鲁肽对阿托伐他汀、地高辛、二甲双胍、华法林、左炔诺孕酮等口服药物PK的影响。结果发现,与司美格鲁肽联用后,阿托伐他汀的Cmax降低38%,Tmax延迟(从0.74h变为2h),对AUC无影响。对地高辛、二甲双胍、华法林Cmax和AUC均无影响。炔雌醇和左炔诺孕酮的Cmax不受影响,AUC有增加趋势。
虽然司美格鲁肽对部分药物PK有一定影响,但无临床意义,比如阿托伐他汀的药效与Cmax相关性不强。因此,联用药物也无需调整剂量。
6)指南:除非临床前或临床观测到心律失常风险,一般不需要开展QT间期研究
司美格鲁肽52周BIW食蟹猴毒理研究中发现高剂量1只雌性动物出现了慢性左束支传导阻滞,该剂量对应的暴露量是临床拟用剂量的27倍。虽然心束支传导阻滞在猴和人中是偶发的,且大多数情况下与其它心脏疾病相关,但与司美格鲁肽的相关性不能完全排除。临床研究期间,开展了司美格鲁肽的QTc研究,剂量设定为0.5、1、1.5mg/人,未发现QT间期延长的风险。因此,司美格鲁肽的QT研究思路与临床药理学指南要求倒是一致的。