1995年,美国食品药品监督管理局(FDA)提出了生物药剂学分类系统(BCS)[1],即一种科学框架、将药物基于其溶解性和渗透性分为 4 类,目的是为药品扩大生产规模、上市后的变更(SUPAC)授予生物等效性(BE)豁免,并以溶出度试验作为判定变更前后是否等效的依据,来替代昂贵耗时的体内研究[2-3]。当时 BE 豁免仅用于药品的变更申请。随后,BE豁免的范围被扩大至某些口服仿制药的上市批准。FDA于2000年颁布的《基于生物药剂学分类的速释口服固体制剂体内生物利用度和生物等效性豁免的指导原则》,仅活性成分为高溶解性、高渗透性的药物(BCS I类)且为口服速释制剂的产品能被豁免,在 2017年颁布的更新版本[4]中进一步将适用范围扩展至活性成分为高溶解性、低渗透性的药物(BCS III类)。BE豁免的主要优势在于可以简化药品的批准流程、减少时间,从而降低了上市成本。
经检索中国国家药品监督管理局(NMPA)及其他权威监管机构[如FDA、欧盟药品管理局(EMA)、世界卫生组织(WHO)]出台的有关人体 BE 豁免的指导原则,可将 BE 豁免大致分为 2 种类型:第 1 种是基于仿制制剂某一规格产品的 BE试验显示与参比制剂生物等效去豁免其他规格的BE试验;第2种是基于 BCS 分类豁免某种药品(所含的原料药为BCS I类、BCS III类)的BE试验。
针对上述 2 种类型的豁免,中国 NMPA 已出台了《以药动学参数为终点评价指标的化学药物仿制药人体BE研究技术指导原则》[5(] 第1种豁免)、《人体生物等效性试验豁免指导原则》[6(] 第2种豁免),FDA 颁布了《工业指南草案:按简化新药申请提交的以药代动力学为终点的生物等效性研究》[7]、《基于 BCS 分类系统的速释口服固体剂型的体内生物利用度和生物等效性研究》[4],其中中国的指导原则主要参考FDA的制定;而EMA、WHO则将第1种和第 2种类型整合在 1个指导原则中,即《生物等效性研究指南(2010 年版)》[8]、《WHO 技术报告系列第37 号附件 7:仿制药实现其可替代性的注册要求指南》[9]。
为协调统一各成员国监管机构的技术要求,国际人用药品注册技术协调委员会(ICH)于 2019 年针对第 2 种豁免颁布了《M9:基于生物制药分类系统的生物豁免指导原则》[10],并在问答文件中阐述了该指南允许在仿制药申请中存在地区性差异,以适应某些现有法规不允许仿制药申请采用基于BCS豁免BE的例外情况。中国NMPA及美国FDA均陆续发布了实施该指导原则的通告,以替代原有的指导原则。EMA 在官网发布了 ICH M9 并表明生效日期为2020年7月3日,但同时强调基于BCS I和 III 类豁免 BE 试验并未在全球内达成共识,申报者需遵循不同地区的法规。日本药监局(PMDA)于2020 年 12 月在官网上公开了 M9 日文稿[11]并表明该指南可适用于部分申请(因参比制剂的处方难以获得,暂不适用于仿制药)。综上,ICH M9 基本替代了各国原有的第2种BE豁免指导原则。而对于第 1 种豁免,目前 ICH 正在起草并计划于 2024 年完成《M13:速释口服固体制剂的生物等效性》的制定,有望对各国现有的不同指导原则进行协调统一。
上述指导原则适用范围存在差异之处,但均包括了速释口服固体制剂,WHO、PMDA 还明确了第1 种豁免亦适用于其他的特殊制剂(如迟释、缓释等)及非口服给药途径药物,FDA、ICH M9提出第2种豁免还适用于新药开发。因篇幅有限,本文分析的对象仅针对申报量大的速释口服固体剂型仿制药,旨在通过比较国内外相关指导原则对速释口服固体剂型的仿制药药学研究的要求,重点关注存在的差异之处并在最终经 ICH 协调一致的过程中得到思考和启示,了解不同监管机构的申报要求,以期使得研究者少走弯路,增加仿制药被豁免体内试验的成功率、进而降低开发成本,但同时亦能保证其质量和疗效与参比制剂一致,真正实现其临床可替代性。
一、参比制剂的选择
在仿制药的注册申报中,均须以参比制剂为标杆进行相关的研究,因此需合理选择参比制剂,而关于参比制剂研究批次的选择,不同监管机构的要求如下:
(1)日本《仿制药BE研究的指导原则》[12]规定,取3批原研制剂进行溶出曲线研究,在其规定的溶出方法下测定,选取中间那条溶出曲线的批次作为参比制剂样品,若 3 批原研制剂在 15 min 内溶出度均能达到 85%,则任一批次均可作为参比试剂,用于 BE批的参比制剂含量需与标示量接近且与仿制制剂含量的差值最好不超过 5%;
(2)中国要求多批[13(] 通常为 2 批及以上)进行药学对比研究,建议BE 批参比制剂含量与仿制制剂含量的差值小于5%[5];
(3)EMA 明确用于 BE 试验参比制剂的选择应基于含量(与仿制制剂的含量差值应不超过 5%)和溶出数据并要求申请人提供资料说明如何基于溶出和含量选择代表性批次的参比制剂,建议研究1 批 以 上 的 参 比 制 剂 以 确 定 BE 试 验 用 的 批次[8];
(4)ICH M9征求意见稿中明确了至少 1批,而在正式稿中则删除了关于参比制剂批次的描述[10]。
二、对原料药的具体要求
第2种BE豁免(基于BCS分类),对原料药本身的理化性质(如结构、溶解性)进行了限定。
其中关于溶解度的测定各指导原则均明确应在pH值1.2~6.8范围内的水溶性介质中进行,EMA和 ICH 还分别说明若药物的 pKa、最低溶解度所对应的 pH 值在 1.2~6.8 范围内,则需评价相应 pH 值下的溶解度,而我国及FDA指导原则还明确需考察pH=pKa±1 处的溶解度。对于测试药物的用量,我国、EMA、WHO及ICH指导原则均明确采用最高剂量进行试验,ICH M9 还提出若单次治疗的最高剂量不符合高溶解性标准,但参比制剂的最高规格在要求条件下可溶解,则应提供额外数据以证明基于BCS分类豁免BE的合理性。
各指导原则中,ICH M9 最为详细地阐述了溶解度的测定方法,强调了研究者容易忽略的试验细节问题,如:
(1)溶解度试验时间:应证明溶解度在预期吸收时间范围内(在问答文件中说明了如何确定溶解度测定的持续时间)能维持稳定。
(2)应在添加药物活性成分后和平衡溶解度研究结束时测定每种试验溶液的 pH 值,以确保溶解度测定是在指定pH值下进行,必要时应调节pH值。
(3)将以pH值1.2~6.8内测得的最低溶解度对药物活性成分进行分类。
(4)应采用经适当验证的方法测定溶解度,使用药典中适当的介质,在每个溶解度条件或 pH 值下至少平行测定3个样本。
(5)应证明药物活性成分在溶解介质中具有足够的稳定性,若药物活性成分在溶解度测定过程中不稳定,即降解>10%,则不能充分确定其溶解度,因此无法分类。
在M9问答文件中明确应提供试验获得的溶解度数据以确定药物活性成分的溶解性。
三、对制剂处方工艺的要求
3.1 第1种豁免
基于某一规格生物等效豁免其他规格 BE 试验,其他规格的处方工艺则须满足一定的前提条件,下文介绍不同监管机构的具体要求。
3.1.1 NMPA[5]
若申请仿制制剂的其他规格 BE豁免,则各规格制剂的处方比例相似,是指以下情况:
(1)不同规格之间所有活性和非活性组分组成比例相似(2022年1月21日发布的问答文件[14]对此种情况进行了解释说明)。
(2)对于高活性(highpotency)的药物(活性成分在制剂中所占质量比例低):①不同规格的制剂质量一致(差异不超过10%);②各规格使用相同的非活性组分;③规格的变更系通过改变活性组分的用量以及1个或多个非活性组分的用量来实现。
3.1.2 FDA[7]
处方相似性判断有 3种情况:(1)不同规格之间所有活性和非活性组分组成比例相似,如50 mg规格所有非活性成分为100 mg规格的1/2,或为 25 mg 规格的 2 倍;
(2)符合以下条件:①不同规格的制剂质量几乎相同(与 BE 规格的差异不超过10%);②各规格使用相同的非活性组分;③规格的变更系通过改变活性组分的用量以及1个或多个非活性组分的用量来实现。
(3)不同规格间活性和非活性成分比例不相似但有充足的证据可证明处方相似的(FDA将在ANDA评估期间对处方相似性进行认定)。
3.1.3 EMA[8]
(1)相同的制备工艺;(2)不同规格处方定性组成相同;(3)不同规格处方组成比例相似,如所有规格的原辅料比例相同(对于速释制剂,包衣成分、胶囊壳、色素和香精除外)。当处方等比存在一定误差时,若满足以下①和②或①和③时则仍可判定处方相似,即可考虑豁免 BE:①活性成分占片芯或胶囊内容物质量的比不超过 5%;②片芯或胶囊内容物中辅料的量与 BE 规格相同,仅活性成分的量发生变化;③填充剂的改变量=活性成分的改变量,其他辅料的量与BE规格相同。
3.1.4 WHO[9]
不同规格制备工艺相同且处方相似,以下 2 种形式均可被认为处方相似:
(1)不同规格之间所有活性和非活性组分组成比例相似(如 50 mg规格片剂中含有的活性和非活性组分均为 100 mg规格片剂的 1/2 或者为 25 mg 规格片剂的 2 倍),对于速释制剂,包衣成分、胶囊壳、色素及香精通常无上述要求;
(2)对于制剂中原料含量相对较低的情况(单位剂量最多含 10 mg,或质量百分比不超过5%),所用规格间的单位质量相似,若满足以下条件则可考虑豁免:①片芯或胶囊内容物中辅料的量与BE规格相同,仅活性成分的量发生变化;②填充剂的改变量=活性成分的改变量,其他辅料的量与BE规格相同。
由上可知,除 FDA 外,各监管机构对于处方相似性的判断均分为两种情况。第 1 种情况,即不同规格间原料药和辅料用量均呈等比增加或减少;而第 2 种情况,各指导原则主要对原料药百分比含量的要求略不同,EMA 明确为占比不超过 5%,WHO在 EMA 的基础上增加了另一种情况(即单位剂量含有的活性成分至多10 mg),中国于2022年1月21日发布“对我国《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》中关于多规格豁免 BE药学评价标准“处方比例相似性”相关问题的问答(试行)”[14],在该文件中明确了高活性药物的标准为占比<5%。另外,EMA及 WHO 均要求不同规格与 BE 批次有相同的制备工艺。
EMA、WHO 还提出当多个规格间不符合处方相似的要求时,可采用括号法,这样仅进行两端的两个规格(如最高和最低规格,或处方组成差异最大的 2 个规格)的体内 BE 试验即可,则剩余规格组成上的任何差异均可被上述两个试验覆盖。
3.2 第2种豁免
3.2.1 基本原则
无论基于 BCS I 类还是 III 类豁免 BE试验,均对辅料的选择有所要求。ICH M9提出,理想情况下,受试制剂的辅料组成应模仿参比制剂。但是,若辅料存在差异,应评价这些差异可能对体内吸收产生的影响。评价中应考虑药物活性成分性质和辅料作用。对于片剂包衣中的较小用量或低于对特定药物活性成分产生影响的已知阈值的用量,可相对较少关注。
需注意的是,不管是 BCS I 类还是 III 类,ICHM9均规定可能影响吸收的辅料应种类相同且用量相似,即在参比制剂辅料用量的±10% 范围内且这些辅料的累计差异应在±10% 范围内。对于 BCSIII 类药物而言,除了对影响吸收的辅料有要求外,处方中其他辅料的种类也应相同、用量相似。
WHO 强调若某药品中 1 个规格基于 BCS 豁免BE 试验,则其他规格也必须基于 BCS 分类来评估是否可以豁免,而不是基于处方等比;ICH还明确必须每个规格均与参比制剂比较来支持基于BCS分类豁免BE。
3.2.2 处方相似性判断
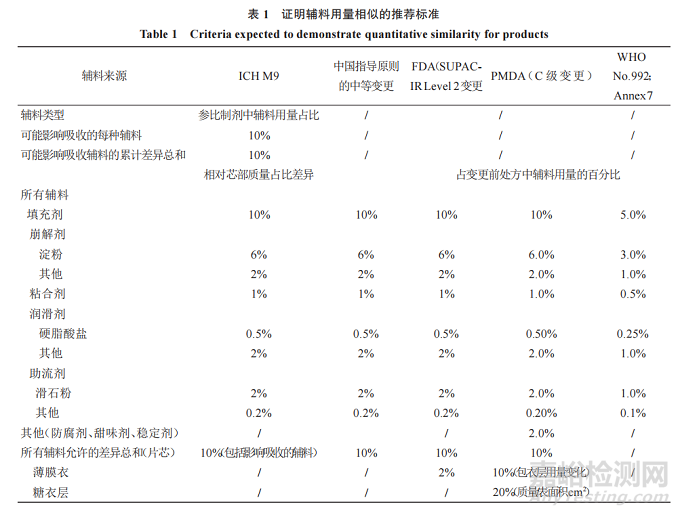
对基于 BCS分类豁免 BE试验的各指导原则中对处方相似的判断标准及各变更指导原则中申请豁免 BE对处方辅料用量变化的可接受范围进行列表分析(表1)。FDA基于BCS分类豁免 BE试验的指导原则是由其变更指导原则延伸而来的,其处方相似性的判断标准同 SUPACIR Level 2[2-3]的要求,中国现行变更指导原则的中等 变 更[15]、ICH M9 及 PMDA[16-17]的 相 关 规 定 与FDA 大体是一致的(除对包衣的要求不同外)。而WHO[11]的标准最为严格,除未对包衣膜进行限定外,其他同 FDA SUPAC-IR Level 1,亦与我国的微小变更可接受范围一致。
ICH M9 问答文件[10]解释:芯部各辅料百分比差异是以相对仿制制剂与参比制剂各自的芯部进行计算,若仿制制剂符合这些标准,但辅料绝对质量存在很大差异(如仿制制剂与参比制剂的芯部质量不相似),可能须额外论证。
FDA变更指导原则指出,发生Level 2变更的药物属于高溶高渗(BCS I 类)、低溶高渗(BCS II 类)或高溶低渗(BCS III类)时,若溶出度等满足条件可不进行 BE 研究,否则应归属于 Level 3,需进行 BE试验(或证实体内外具有相关性时可能会考虑豁免BE)。
中国现行变更指导原则规定的中等变更与FDA 的 Level 2 变更相比,除未限定薄膜衣的差异外,其他均一致。而中国先前颁布的变更指导原则[18]对包衣液用量变更进行了解释,即:以原处方单剂量理论质量计算,一般允许变更幅度为±2%且包衣液组成不能变化。PMDA 提出对于 C 级变更,在药物的溶解性和治疗窗满足规定时,若溶出等效则可视为生物等效 ,否 则 需 根 据 仿 制 药 BE 研 究 指 南[12]进 行 BE试验。
总体上来看,对于处方相似性的判断,不同指导原则主要对包衣膜的要求存在不同,ICH M9 在FDA 指导原则的基础上,删除了包衣膜用量的控制,可能因为 BCS I和 III分类药物制备的速释制剂溶出均较快,受包衣膜影响的可能性较小,且包衣用量的准确测定亦存在一定困难。而日本变更指导原则最大的不同在于其额外明确了含量较少的一些辅料(如防腐剂、甜味剂、稳定剂等)的变化量要求,并分别对薄膜衣和糖衣的组成和包衣质量/片芯表面积的变化进行了明确规定(变化水平将片芯和包衣层分开计算),将包衣层中辅料独立计算的原因在于某种情况下包衣层影响药品的溶出曲线,应考虑包衣的厚度而不是包衣层的质量,因此在指定包衣层可接受变化的标准时有上述 2 个指标(包衣层组成变化和包衣质量/表面积比的变化)。
另外,对于辅料不同等级(或型号)问题,ICHM9问答文件中说明:若适合,应基于制剂中辅料的功能特性来评价辅料等级的差异。对于一些辅料,等级改变可能会影响药物制剂的溶出(如 HPMC粒度分布、黏度和取代度的改变;硬脂酸盐润滑剂比表面积的改变)。对于辅料相似性的评价,需要具体情况具体分析,以证明“种类相同”。
关于处方相似性的计算方法,ICH M9 及日本指导原则中均给出了案例解析。
四、对溶出曲线的要求
4.1 第1种豁免
各指导原则均明确要求不同规格制剂的体外溶出曲线相似,EMA 规定溶出介质为 pH 1.2、4.5、6.8,WHO 额外增加了质量控制(QC)介质(被药典收载的),各介质中溶出均需相似(除非经论证不满足漏槽条件)。当不同规格因在上述介质中不满足漏槽条件导致溶出不相似时,需提供进一步研究资料来证实,如通过测试相同剂量的样品(如2个5 mg的片剂 vs 1个 10 mg 的片剂)得出的溶出具备相似性 ,或经试验表明参比制剂具有相同的溶出行为。
4.2 第2种豁免
各监管机构曾实施的指导原则均对溶出曲线有具体要求,ICH M9 主要在以下几个方面进行了协调统一。
(1)溶出介质种类 :另增了 1 个要求 ,即“需在最低溶解度 pH(若与规定的 3 种缓冲液不同)下进行额外研究”,同时强调了不应使用有机溶剂和添加表面活性剂。
(2)溶出介质体积:中国及FDA曾实施的指导原则分别为500 mL或更少、500 mL或更少(经论证后可选择 900 mL),ICH M9 统一为“900 mL或更少(建议使用质控(QC)检测所选择的体积)”
(3)桨法转速:中国、FDA及WHO允许一定条件下采用75 r·min−1,而ICH M9参照EMA的要求,仅允许采用50 r·min−1,并在M9问答文件中提到BCS I 类药物预计不会发生高变异的情况,若发生变异、堆积、黏附、漂浮等现象 ,建议经论证后采用篮 法 100 r·min−1 测 定 ,而 未 说 明 可 以 提 高 桨 法转速。
关于溶出相似性评价,各国指导原则的要求均相同。BCS I 类药物,仿制制剂与参比制剂需具有快速溶出(30 min 内 API 的平均溶出百分比均能≥85%)的特点且相似(基于f2比较)。BCS III类药物,仿制制剂与参比制剂需具有非常快速溶出的特点,即在 15 min 内 API 的溶出均能达到标示量的 85%以上。对于超过 1 种规格的制剂,仿制制剂和参比制剂的每个规格均应进行溶出曲线比较。
ICH M9问答文件中还明确,对于BCS I类药物预计不会发生溶出存在高变异的情况,因此,不适宜采用替代统计方法证明相似性(如boost strapping法)。若因堆积效应产生高变异,经科学论证,可考虑替代方法以解决堆积效应等问题。溶出曲线相似性评价时,应使用仿制制剂和参比制剂的12个制剂单位所得的溶出数据,报告每个独立的制剂单位在每个特定的时间点的溶出量(以标示量的百分数表示),将平均溶出量 ,溶出范围(高低值)和变异系数(相对标准偏差)列表呈现;在进行 f2因子计算时,采样点及相对标准偏差值需满足要求。
五、结语
在 ICH 文件颁布前,不同监管机构有关 BE 豁免的指导原则技术要求均存在一定的差异,本文主要对上述几个关键药学问题进行了探讨。药品研发时重点关注不同之处,积极进行相关试验研究,做出最科学严谨的选择。
5.1 参比制剂的选择
各监管机构均明确参比制剂与仿制制剂的含量差值不超过 5%,日本额外强调了参比制剂的含量需与标示量接近;而对参比制剂批次的要求不尽相同,但基本上均建议对多个批次的参比制剂进行研究。ICH M9终稿中未对参比制剂的批次进行明确,可能是考虑到各国监管机构执行层面的差异,未作统一规定。若发现参比制剂的溶出等存在较大的批内或批间差异,建议增加批次研究,对其质量有全面的了解,以选择合理的批次。
5.2 处方工艺的要求
5.2.1 处方相似性的判断
通过对比不同监管机构的指导原则及最终由 ICH 协调统一的过程中可以发现,基于 BCS 分类申请豁免 BE 时辅料用量的可接受标准是由早期 FDA 颁布的变更指导原则发展而来的,即基于近 30 年的实践积累,而非一蹴而就。日本界定药品的变更等级时,分别对包衣膜组成和包衣质量与芯部表面积比进行了明确规定,并给出了考察包衣对溶出曲线影响的方法(如采用水杨酸空白片进行试验)。研究者可结合参比制剂的实际包衣情况(如包衣组成、增重及包衣质量与片芯表面积比),考察包衣对溶出曲线可能存在的影响,同时结合药品自身稳定性特点(对光湿等的敏感性)最终确定合理的包衣组成及增重,如对于光不稳定的药物,包衣的组成(是否含有遮光剂)及增重对强光下的稳定性影响较为显著;对于易吸潮的药 物 ,包 衣 的 主 要 膜 材 、增 重 及 致 密 性 均 可 能影响片芯对水分的吸收程度。
ICH M9问答文件表明,2个制剂中辅料的绝对质量应不存在很大差异,即芯部质量需具有相似性,但未明确具体标准,FDA在第1种豁免指导原则中提出片芯质量差异在±10% 以内认为处方相似,因此 10% 的标准可作为参考依据之一,但基于 M9中提出的理念“理想情况下,受试制剂的辅料组成应模仿参比制剂。”,建议研究者尽量选择与参比制剂相近的芯重。亦如 WHO 提到的,对于仿制药中辅料的选择,统一的原则就是,越接近参比制剂,越容易豁免 BE 试验。在研发早期,可以检索各监管机构参比制剂的审评报告、说明书及相关专利等以获得全面详细的处方信息,或者运用一定的物理化学方法逆向解析参比制剂的处方,特别是可能影响溶出的辅料,为了获得其准确用量,建议对分析方法进行必要的验证。
5.2.2 生产工艺
针对第一种豁免,EMA 及 WHO均要求不同规格与 BE批次有相同的制备工艺。为避免不同的工艺或关键工艺参数可能导致的体内不等效风险,建议不同规格间采用相同的工艺,或参考中国现行的指导原则进行相关研究。
5.3 溶出曲线
文中所述的QC方法,WHO解释为已被药典收载的方法,通常为有一定鉴别力的溶出方法,EMA相关指导原则[19]对鉴别力的溶出方法定义为“方法可区分采用不同关键工艺参数和(或)关键物料属性(可能对生物利用度有影响)所生产的不同批次样品的能力。理想上所有不等效的批次均可被体外溶出方法监测出”。
基于BCS分类豁免BE对溶出曲线测定条件的要求中须注意到,ICH M9 另增加了 1 个 QC 介质体积及1个最低溶解度pH值下的介质种类(若与规定的3种缓冲液不同),且不可采用桨法75 r·min−1。
另外,在中国若新增药品规格,需按补充申请的途径申报,药审中心于 2022 年 11 月发布《〈已上市化学药品药学变更研究技术指导原则(试行)〉中溶出曲线研究的问答》[21],给出了新增规格溶出曲线研究的具体建议。
本文分析不同监管机构先前实施的法规与最终协调一致的ICH 指导原则存在的差异,了解指导原则的起草、颁布及后续不断完善的整个过程,更能深刻的理解其背后的科学依据,对研发高质量药品起到事半功倍的作用。同时提醒申请人注意,ICH 仅是在技术层面进行协调统一,其允许在仿制药申请中存在地区性差异(如日本因参比制剂处方难以获得、暂不适用于仿制药),EMA明确基于BCS分类的豁免尚未在全球内达成共识,申请人须遵循当地法规。