您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-12-08 22:36
本文根据借鉴了法规的要求,同时结合笔者的工作经验 ,整理此稿供交流学习。
随着《美国药典》USP新通则<232>和USP<233>、ICH Q3D元素杂质指南、《中国药典》元素杂质限度限度和测定指导原则等法规的实施日益受到药企重视,同时元素杂质研究也作为关系到药品国内外注册申报、原料药等进出口贸易直接相关材料。所以本文从元素杂质的风险评估、不同法规对元素杂质的要求、以及分析方法开发与验证三个部分分享元素杂质对医药相关企业思考。
一、元素杂质的风险评估
风险评估过程可以描述为以下三步:(1)识别已知和潜在可能进入药品的元素杂质来源(2)通过测试已知或预期杂质,将其水平与已有PDE 值比较,评估药品中特殊的元素杂质出现的可能性,(3)总结和记录风险评估。识别出工艺中嵌入控制是否充分,或识别出要考虑增加控制来限制药品中的元素杂质
在很多情况下,这些步骤其实是同步的。风险评估的结果,可以是一个迭代的结果,用以建立一种方法来保证潜在元素杂质不超过PDE 值。
元素杂质从来源上说,一般由四个来源,(1)生产过程中有意添加的元素(例如催化剂)的残留杂质;(1)是非有意添加, 但原辅料、生产用水等引入的元素杂质;(3)生产设备引入的元素杂质;(4)是由容器密封系统浸出至API或制剂的元素杂质。所以我们在原材料的选购、生产设备的选择、药品内包材的使用、一次性生产使用系统使用过程中,都要考虑到元素杂质的引入风险。
在对潜在元素杂质进行识别后,可能会有两种结论:(1)风险评估未能识别出任何潜在元素杂质。风险评估的结论和支持性资料和数据要进行记录。(2) 风险评估识别出了一个或多个潜在元素杂质。对所有识别出的元素杂质,如果该元素杂质有多个来源,要考虑进行风险评估。并记录评估的结论和支持性资料。
总结要考虑相对于元素杂质PDE 值,已观察到的或预期的元素杂质水平的显著性。这里,将药品中已建立的PDE 值的30%定义为控制阈值,作为对已观察到的元素杂质水平的显著性的衡量方法。
如果药品中所有来源的总元素杂质水平预期会保持一致,且低于PDE 值的30%水平,而申报人已对数据进行了适当的评估,证明对元素杂质的控制已经足够充分,则不需要采用更多的控制措施。
如果风险评估未能证明一种元素杂质的水平具有一致性且低于控制阈值,则要建立控制措施来保证元素杂质水平不会超过药品的PDE 值。
控制元素杂质是药品全面控制策略的一部分,它能保证元素杂质不超过PDE 值。当元素杂质有可能超过控制阈值时,需采取更多措施来保证其水平不会超过PDE 值。申报人可以采用的措施包括但不仅限于:(1)改进生产工艺步骤,通过特定或非特定的精制步骤将元素杂质降低至控制阈值以下;(2)实施中控或上游控制,用以将元素杂质的浓度限定在制剂的控制阈值以下;(3)建立辅料或原料的质量标准限度(例如,合成中间体)(4)建立原料药质量标准限度;(5)建立制剂质量标准限度;(6)选择适当的容器包装系统。
二、不同法规对元素杂质的要求及控制方法
(1) EMA、EP关于元素杂质的要求
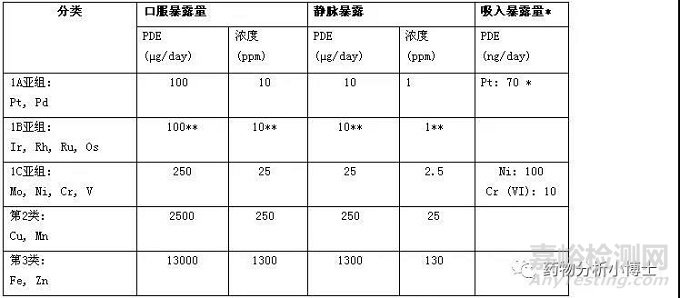
2008年2月21日EMEA/CHMP颁布了金属催化剂或金属试剂残留量限度规定的指导文件(GUIDELINE ON THE SPECIFICATION LIMITS FOR RESIDUES OF METALCATALYSTS OR METAL REAGENTS),并将于2008年9月1日在欧盟正式实施。该指导文件从1998年6月开始起草,历经多次咨询、讨论,最后定稿。该指导文件基于安全考虑(对人体健康的潜在风险),将金属分为以下3类。
第1类金属:具有显著安全性担忧。这一类金属具有已知的或怀疑的人体致癌性,或者具有其他显著的毒性。包括铂(Pt)、钯(Pd)、铱(Ir)、铑(Rh)、钌(Ru)、锇(Os)、钼(Mo)、镍(Ni)、铬(Cr)、钒(V)十种金属。其中第1类金属又被分为1A、1B 、1C三个亚组,1A亚组包括Pt、Pd。1B亚组包括Ir、Rh、Ru、Os。1C亚组包括Mo、Ni、Cr、V。
第2类金属:具有低的安全性担忧。这一类金属对人体有潜在的较低毒性。通常可以较好耐受此类金属在常见药物中的暴露量。可能是营养需要的痕量金属,常存在于食物原料中或营养补充剂中。包括铜(Cu)、锰(Mn)。
第3类金属:安全性担忧最小。这一类金属无明显毒性。已建立了安全范围,在远大于常见药物中的量时,也可以较好耐受。通常广泛存在于环境、植物或动物中。包括铁(Fe)和锌(Zn)。
该指导文件的14种金属分类见表1。
表1:金属催化剂和金属试剂的分类、PDE和浓度限度
(2) ICH Q3D元素杂质的要求
ICH于2009年10月批准了Q3D,经过多方讨论后,修订版本的Q3D step4于2014年12月16日生效,其中列出了24种元素杂质的三种给药途径的PDE值,确定了实施日期为:新上市许可为2016年6月生效,已上市品种为2017年12月生效。
ICH Q3D规定了元素杂质在药品、原料药和辅料中的允许浓度,单位为ug/g。可以使用这些浓度来评估日剂量不超过10g/天的药品中含有的元素杂质。数值如下表。
|
元素 |
分类 |
口服浓度 |
注射浓度 |
吸入浓度 |
|---|---|---|---|---|
|
Cd 镉 |
1 |
0.5 |
0.2 |
0.2 |
|
Pb 铅 |
1 |
0.5 |
0.5 |
0.5 |
|
As 砷 |
1 |
1.5 |
1.5 |
0.2 |
|
Hg 汞 |
1 |
3 |
0.3 |
0.1 |
|
Co 钴 |
2A |
5 |
0.5 |
0.3 |
|
V 钒 |
2A |
10 |
1 |
0.1 |
|
Ni 镍 |
2A |
20 |
2 |
0.5 |
|
Tl 铊 |
2B |
0.8 |
0.8 |
0.8 |
|
Au 金 |
2B |
10 |
10 |
0.1 |
|
Pd 钯 |
2B |
10 |
1 |
0.1 |
|
Ir 铱 |
2B |
10 |
1 |
0.1 |
|
Os 锇 |
2B |
10 |
1 |
0.1 |
|
Rh 铑 |
2B |
10 |
1 |
0.1 |
|
Ru 铷 |
2B |
10 |
1 |
0.1 |
|
Se 硒 |
2B |
15 |
8 |
13 |
|
Ag 银 |
2B |
15 |
1 |
0.7 |
|
Pt 铂 |
2B |
10 |
1 |
0.1 |
|
Li 锂 |
3 |
55 |
25 |
2.5 |
|
Sb 锑 |
3 |
120 |
9 |
2 |
|
Ba 钡 |
3 |
140 |
70 |
30 |
|
Mo 钼 |
3 |
300 |
150 |
1 |
|
Cu 铜 |
3 |
300 |
30 |
3 |
|
Sn 锡 |
3 |
600 |
60 |
6 |
|
Cr 铬 |
3 |
1100 |
110 |
0.3 |
同时,如果药品中元素杂质允许浓度不适用时,指导原则也给出了方法2a、2b或3的允许浓度限值的计算方法。
(3) USP<232>、USP<233>元素杂质的要求
USP<232>、USP<233>元素杂质的要求从元素杂质种类和限值要求上基本与ICH保持一致,USP<233>推荐了两种基于现代分析仪器额检测方法,方法1是ICP-AES法(或ICP-OES法)、方法2 是ICP-MS,这两种分析方法具有较高的灵敏度和准确性,可以检测出药品中很低残留量的元素杂质。同时,针对固体样品,新通则给出了首选密闭容器微博消解技术作为固体样品的前处理方式。
(4)中国药典对重金属元素杂质的要求
中国药典2015版 四部通则 0821重金属检查法 对重金属检测的检测方法仍然使用比色法。2017年随着中国食品药品监督管理局加入ICH成员,未来中国的药品法规也是向ICH指南靠拢。所以2018年国家药典委员会发布了《元素杂质限度和测定指导原则》2020年版第一次征求意见稿。元素杂质研究要求正式接轨ICH Q3D的要求。
三、元素杂质分析方法开发与验证的经验分享
(1)仪器设备的选择
目前比色法依然是中国药典采用的方法,但因为这一方法无法准确定性定量不同重金属元素的含量,缺乏专属性、灵敏度的要求,所以检测方法限定为ICP-MS法、ICP-OES法(或者ICP-AES法)、原子吸收分光光度法。笔者实验室均有这些仪器设备,所以分析一下各仪器设备的优劣势供大家参考。
ICP-MS法:
优势:检测限低至ppt级别,线性范围宽,专属性强, 多元素同时检测;
劣势:光谱干扰严重,耐盐性差,钾钙钠等元素干扰严重,仪器成本高。
ICP-OES法(或者ICP-AES法)
优势:检测限高于ICP-MS,精密度更准确度高,线性范围宽,多元素同时检测
劣势:谱线复杂、干扰强烈、导致分析测试能力下降。
原子吸收分光光度法(FAAS GFAAS)
优势:常用于Na、K、Ca、Mg、Fe、Zn、Li等元素的分析。仪器不贵,重现性好。
劣势:基质干扰严重、线性范围窄、只能一次检测一个元素,时间成本高。
所以我们在原料药中进行方法学开发仪器选择时,需要根据目标物的种类和个数来选择仪器设备。常规ICH Q3D中24元素基本选择ICP-MS来操作最为方便,目标物限值浓度较高时建议选择ICP-OES比较合适。选择了仪器设备,我们下一步进行试验部分。
(2)前处理方法的选择
明确了要分析的金属元素之后,就要确认方法开发与验证中最重要的部分-样品前处理。好的方法,从前处理开始。与指导原则不同,USP<233>对于适用的四种前处理方法做出了明确的规定。需要强调的是,绝大多数的验证数据偏差和后续方法开发的挑战,都来自前处理的环节。虽然每种方法都有自己的特点和适合的样品类型(见下表),总体来说直接溶解法在操作的简易性与安全性,结果的稳定性上都具有一定的优势,也是可以最先尝试的步骤。
四种前处理方法特点对比如下:
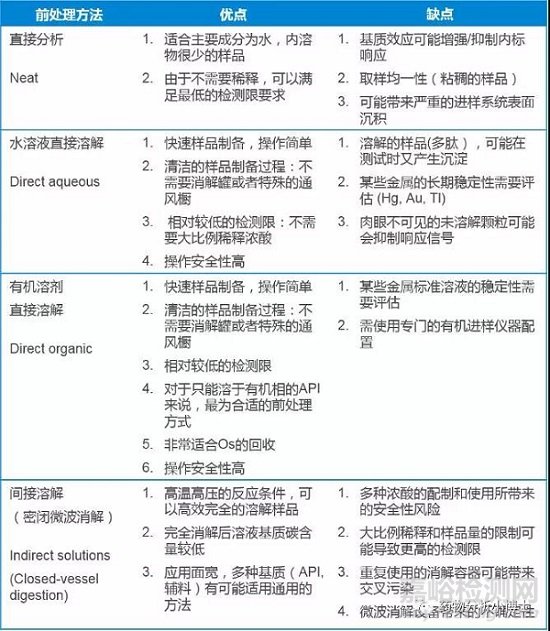
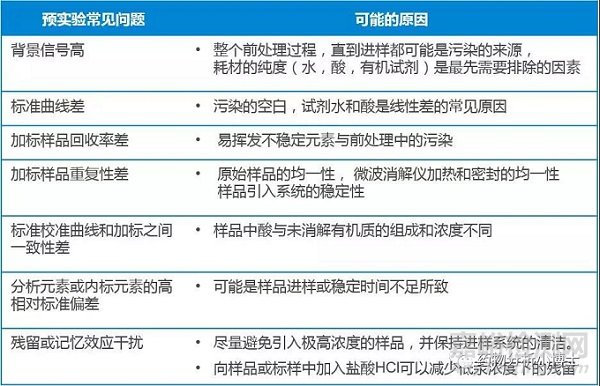
对于有些易于直接溶解于有机溶剂(比如异丙醇, 二甲亚砜等等)中的医药中间体来说,由于避免了高温高压的反应过程,很多反应性,挥发性强的金属杂质都能获得不错的回收,可以为后期的方法验证以及最终的QC测试节约大量的工作量。 对于不能直接溶解的化合物,还可以选择间接溶解法。其中密闭容器微波消解法是最主流的选择,涉及的方法参数组合也比直接溶解要复杂些。选择消解方式时需要考虑酸的选择、样品量的选择、消解模式程序的选择问题。(3)预实验在摸索出完全溶解样品的方法之后,需要进行预实验确认消解溶液的的稳定性,以及设置仪器参数的准确度和精密度的大致范围,为最终的验证做准备。
元素分析预实验常见的问题与原因供参考如下:
(4)完整的方法学验证方法学验证在USP、EP、中国药典中会有区别,但是区别不大。以下按照中国药典2015年版 四部通则 9101为例说明要验证的部分。验证部分一般包括:专属性、线性与范围、本底测试、准确度、重复性、中间精密度、检出限、定量限、耐用性、系统适用性、溶液稳定性等内容。以下简短分别阐述做法。
线性与范围:ICP-MS线性范围广,常规定量分析做0.5J与2J即可,也可作5标准点。检测限与定量限:在制作好标准工作曲线后,连续测定空白溶液21次,记录空白溶液的响应值。根据公式计算仪器检出限以及仪器定量限。系统适用性:常规试验开始前后分别测试三次J标样浓度, 评价RSD。
准确度:高中低浓度加标回收。
重复性:6份J溶液,计算RSD。
中间精密度:常规换人员、时间分别6份J溶液,计算RSD。专属性:根据元素选择对应的内标物质;汞元素测试可以采用金溶液作为汞的清洗液,消除汞记忆效应。耐用性:一般考虑消解温度、消解时间、泵速、溶液稳定性试验。以上根据经验所写,供大家参考使用。

来源:Internet


