为了推动非临床和临床生物样品分析数据在各监管机构间获得互认,保证分析方法的有效性和可靠性,以及确保上市许可申请审评的可靠性,对生物样品分析方法的完善显得至关重要。此外,为了促进不同区域生物样品分析数据集的相互使用,对生物样品分析方法验证(BMV)的国际协调也显得尤为重要。因此,自从2019年2月人用药品技术要求国际协调理事会(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, ICH)(以下简称“ICH”)发布《生物分析方法验证及样品分析》M10(BIOANALYTICAL METHOD VALIDATION AND STUDY SAMPLE ANALYSIS, M10,以下简称“ICH M10”)征求意见稿以来,经过业界多轮讨论,ICH M10最终于2022年5月正式颁布。随后作为ICH监管成员的中国国家药品监督管理局(National Medical Products Administration, NMPA)(以下简称“NMPA”)也为了推动ICH三级指导原则在中国的平稳落地和实施,发布了关于公开征求ICH《M10:生物分析方法验证及样品分析》指导原则实施建议和中文版的通知;在2022年11月美国食品药品监督管理局(U.S Food and Drug Administration, FDA)(以下简称“FDA”)作为ICH的创始监管成员,也发文采纳M10的建议并发布行业指南。在2022年11月16日,欧洲药品管理局(European Medicines Agency, EMA)(以下简称“EMA”)也作为ICH成员发布了对应的Q&A,本文件已在2023年1月21日生效。同时,NMPA在2023年1月29日也发布了2023年第16号公告,自2023年7月29日起的研究均适用《M10:生物分析方法验证及样品分析》国际人用药品协调会指导原则。
首先,我们来看一下ICH M10正式版的更新内容。其中,重点更新了以下几个方面:
对于生物样品分析方法的定义和范围进行了明确。ICH M10明确了生物样品分析方法的定义,包括了用于测量药物和其代谢产物在生物样品中的浓度的所有分析方法。
对于方法验证的步骤进行了详细的描述。ICH M10指出,方法验证应该包括方法设计、方法优化、方法验证、方法转移以及方法的例行监控等步骤。
对于方法验证的参数进行了明确。ICH M10明确了方法验证的参数,包括选择性、灵敏度、精密度、准确度、回收率、稳定性等。
对于方法验证的接受标准进行了明确。ICH M10明确了每个参数的接受标准,以及在何种情况下需要重新验证。
接下来,我们再来看一下NMPA 2020版《中国药典》9012部分指导原则(以下简称“NMPA 2020版”)和FDA 2018年的(Bioanalytical Method Validation, BMV)(以下简称“FDA 2018版BMV”)指导原则的考察项要求。
NMPA 2020版对于生物样品分析方法的要求主要包括了方法的选择、优化、验证和例行监控等环节。其中,对于方法验证的参数,NMPA 2020版明确了选择性、灵敏度、精密度、准确度、回收率、稳定性等参数的要求,但是对于接受标准的要求并没有明确。
FDA 2018版BMV指导原则对于生物样品分析方法的要求与NMPA 2020版相似,但是在方法验证的参数方面,FDA 2018版BMV指导原则更加详细,包括了线性范围、最低定量限度、精密度、准确度、选择性、稳定性、回收率等参数,并且对于每个参数的接受标准进行了明确。
总的来说,ICH M10正式版与NMPA 2020版和FDA 2018版BMV指导原则在生物样品分析方法的要求方面有很多相似之处,但是在方法验证的参数和接受标准方面,ICH M10正式版更加详细和明确。本文会针对具体考察项要求之间的差异进行解析,更加深入地理解ICH M10生物分析方法验证及样品分析指导原则中的内容。
指导原则和法规指南(NMPA/M10/FDA)的差异化解析
替代基质
针对2.2.1完整验证部分,ICH M10“The choice of surrogate matrix should be scientifically justified. Matrix differences within species (e.g., age, ethnicity, gender) are generally not considered different when validating a method.”。值得注意的是同一物种不同种族如外国人与中国人基质间的差异性比对实验无需再考察。
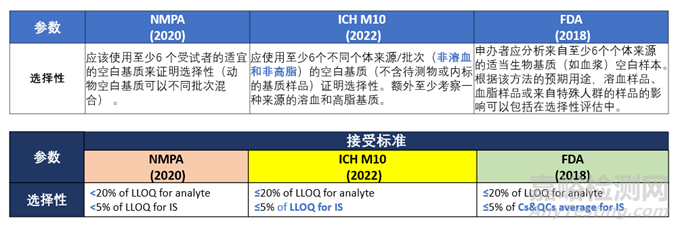
选择性
针对3.2.1选择性部分,ICH M10“Selectivity should be evaluated using blank samples (matrix samples processed without addition of an analyte or IS) obtained from at least 6 individual sources/lots (non-haemolysed and non-lipaemic). For the investigation of selectivity in lipaemic matrices at least one source of matrix should be used. If the drug impacts lipid metabolism or if the intended patient population is hyperlipidaemic, the use of spiked samples is discouraged. This evaluation is not necessary for nonclinical studies unless the drug impacts lipid metabolism or is administered in a particular animal strain that is hyperlipidaemic.For the investigation of selectivity in haemolysed matrices at least one source of matrix should be used. Haemolysed matrices should be obtained by spiking matrix with haemolysed whole blood (at least 2% V/V) to generate a visibly detectable haemolysed sample.”。而NMPA 2020版中的选择性要求,“应该使用至少6个受试者的适宜的空白基质来证明选择性”。FDA 2018版BMV的选择性要求,“Depending on the intended use of the assay, the impact of hemolyzed samples, lipemic samples, or samples from special populations can be included in the selectivity assessment. The sponsor should analyze blank samples of the appropriate biological matrix (e.g. plasma) from at least six (for CCs) or ten (for LBAs) individual sources”。
此部分中ICH M10明确要求至少6个不同个体来源的空白基质中是不包含溶血与高脂基质的,而需要额外考察其选择性,且对于溶血和高脂基质有明确要求,对应的接受标准存在细微差别,详见下表:
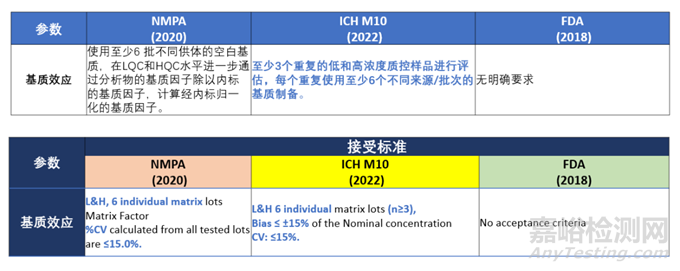
基质效应
针对3.2.3基质效应部分,ICH M10“The matrix effect should be evaluated by analysing at least 3 replicates of low and high QCs, each prepared using matrix from at least 6 different sources/lots. The matrix effect should also be evaluated in relevant patient populations or special populations (e.g., hepatically impaired or renally impaired) when available. An additional evaluation of the matrix effect is recommended using haemolysed or lipaemic matrix samples during method validation on a case-by-case basis, especially when these conditions are expected to occur within the study.”。而NMPA 2020版中的基质效应要求,“使用至少6批来自不同供体的空白基质,通过计算基质存在下的峰面积(由空白基质提取后加入分析物和内标测得),与不含基质的相应峰面积(分析物和内标的纯溶液)比值,计算每一分析物和内标的基质因子,计算经内标归一化的基质因子”。FDA 2018版BMV中无基质效应的要求。
此部分中ICH M10的评估方式与接受标准变动最大,完全改变了常规基质效应中运用基质因子进行评估的做法,因此在进行基质效应的评估时应注意前处理操作方式与接受标准的变化,但不论是ICH M10还是NMPA 2020版都要求除正常基质外,还应关注其他样品的基质效应,例如溶血或高脂,具体操作与接受标准详见下表:
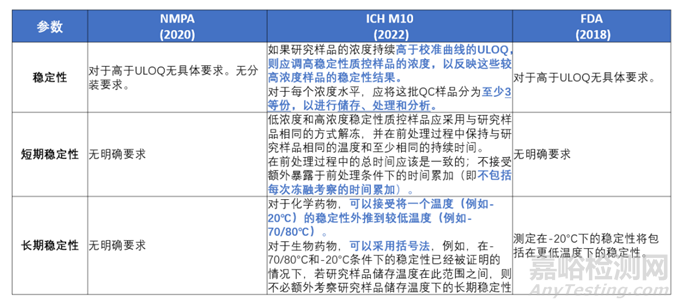
稳定性
针对3.2.8稳定性部分,ICH M10“Stability of the analyte in the matrix is evaluated using low and high concentration QCs. Aliquots of the low and high QCs are analysed at time zero and after the applied storage conditions that are to be evaluated. One bulk QC should be prepared at each concentration level. For each concentration tested, the bulk sample should be divided into a minimum of 3 aliquots that will be stored, stressed and analysed.If the concentrations of the study samples are consistently higher than the ULOQ of the calibration range, the concentration of the high QC should be adjusted to reflect these higher concentrations.”而NMPA 2020版和FDA 2018版BMV中无高于ULOQ的稳定性要求,也无分装、储存、处理和分析稳定性样品的细节要求。因此在进行稳定性样品的准备及其考察时,应注意上述细节来匹配现行法规的要求。
对于短期稳定性,ICH M10要求,“The total time on the bench top should be concurrent; it is not acceptable to use additive exposure to bench top conditions (i.e., time from each freeze-thaw evaluation should not be added up).”。而NMPA 2020版和FDA 2018版BMV中无短期稳定性总时间计算细则。
对于长期稳定性,ICH M10明确表明,“For chemical drugs, the stability at one temperature (e.g., -20°C) to can be extrapolated to lower temperatures (e.g., -70/-80°C). For biological drugs, a bracketing approach can be applied, e.g., in the case that the stability has been demonstrated at -70/-80°C and at -20°C, then it is not necessary to investigate the stability at temperatures in between those two points at which study samples will be stored.”。这样在实操中,譬如有时由于冰箱长时间不开门,研究样品在保存过程中出现低于冰箱验证的温度范围时,即可评估无影响。
FDA 2018版BMV中也有类似描述,如“Determination of stability at minus 20ºC would cover stability at colder temperatures.”,而NMPA 2020版中并无此要求。针对上述内容的比较详见下表内容,
进样重现性
针对3.2.9进样重现性部分,ICH M10“Reinjection reproducibility is assessed by reinjecting a run that is comprised of calibration standards and a minimum of 5 replicates of the low, middle and high QCs after storage. The precision and accuracy of the reinjected QCs establish the viability of the processed samples.”。而NMPA 2020版和FDA 2018版BMV中无单独的部分描述而是在稳定性部分要求的在低和高浓度质控样品浓度水平进行考察,具体的考察详见下表:
已测样品再分析(ISR)
针对5已测样品再分析(ISR)部分,ICH M10“ISR should be performed within the stability window of the analyte, but not on the same day as the original analysis. If the overall ISR results fail the acceptance criteria, an investigation should be conducted and the causes remediated. There should be an SOP that directs how investigations are triggered and conducted. If an investigation does not identify the cause of the failure, the potential impact of an ISR failure on study validity should also be provided in the Bioanalytical Report. If ISR meets the acceptance criteria yet shows large or systemic differences between results for multiple samples, this may indicate analytical issues and it is advisable to investigate this further. Examples of trends that are of concern may include: All ISR samples from one subject fail. All ISR samples from one run fail. All aspects of ISR evaluations should be documented to allow reconstruction of the study and any investigations. Individual samples that are quite different from the original value (e.g., > 50%, “flyers”) should not trigger reanalysis of the original sample and do not need to be investigated. ISR sample data should not replace the original study sample data”,强调了1)ISR需要在待测物稳定性窗口内进行。2)如果ISR结果失败通过调查后不能确定其失败的原因,则应在分析报告中提供ISR失败对试验有效性的影响评估。3)如果ISR满足接受标准,但在多个样品的结果中显示较大的或系统性误差,这可能表明分析存在问题,建议继续开展调查。而对于个别偏差很大的单个样品不必进行调查。而NMPA 2020版和FDA 2018版BMV中并无上述要求。
ICH M10在ISR部分有更加细致的要求和实操性更好的操作指导,如调查失败时对于结果的评估与汇报
交叉验证
针对6.2交叉验证部分,ICH M10“Cross validation should be performed in advance of study samples being analysed, if possible.Cross validation should be assessed by measuring the same set of QCs (low, medium and high) at least in triplicate and study samples (if available) that span the study sample concentration range (n≥30) with both methods, or in both laboratories.”而对于NMPA 2020版和FDA 2018版BMV中有对于交叉验证的内容和接受标准,但是未对需要完成的样品数量与QC的浓度水平进行规定和要求。
对于接受标准,ICH M10无明确规定,“Bias can be assessed by Bland-Altman plots or Deming regression. Other methods appropriate for assessing agreement between two methods (e.g., concordance correlation coefficient) may be used too. Alternatively, the concentration vs. time curves for study samples could be plotted for samples analysed by each method to assess bias.”。而NMPA 2020版中关于交叉验证则有明确规定了接受标准,“对于质控样品,不同方法获得的平均准确度应在±15 %范围内,如果放宽,应该说明理由。对于试验样品,至少67 % 样品测得的两组数值差异应在两者均值的±20 %范围内。”FDA 2018版BMV要求SOP或者验证方案中定义接受标准,“An SOP or validation plan should define the criteria a priori.”。
待测物同为内源性物质的分析方法
针对7.1待测物同为内源性物质分析方法的准确度和精密度部分,ICH M10“Accuracy and precision should meet criteria specified in Sections 3 and 4 for chromatography and LBAs, respectively. The concentration of the endogenous molecule in the blank matrix may be determined and subtracted from the total concentrations observed in the spiked samples. Only the precision can be determined from the analysis of each unspiked/endogenous QC.”建议使用以下公式计算准确度,
对于稳定性来说,除了需要考察实际生物基质中的真实待测物之外,还应证明替代基质中待测物的稳定性,这部分在实操中也尤为重要。
结语
综上所述,我们详细剖析了ICH M10的相关规定,通过差异化分析的方式了解及明确生物分析检测过程中的具体操作及实施,且随着ICH M10的生效,对于一些特殊情况的处理方式在实操过程中也变得更加容易理解与执行。总而言之,ICH M10作为一个统一的行业标准,整合了各个监管机构的生物分析检测法规指南,使得法规依从性下的生物分析检测工作更易执行。
参考文献
[1] US FDA Guidance for Industry, Bioanalytical Method Validation, May 2018
[2]中国药典2020年版,生物样品定量分析方法验证指导原则,2020年12月1日生效
[3] ICH harmonized Guideline, Bioanalytical Method Validation and Study Sample Analysis (M10), effective 24 May 2022