内容提要:敷料是指用以覆盖伤口的医用材料,其作用是暂时起到皮肤的部分屏障功能(物理屏障),即保护伤口。该类医疗器械产品具有品种繁杂、名目繁多、分类复杂和界定困难的特点。因此,自2017 年8 月31 日发布的新版《医疗器械分类目录》实施以来,出现了一些已注册待延续的敷料类产品涉及到管理类别调整或分类不清晰的问题,有些甚至已经影响到注册证的有效延续。文章主要针对前述的第二类敷料类产品在延续注册技术审评过程中发现的常见问题,归纳并进行探讨,为注册申请人和技术审评部门提供参考。
随着人们对于医疗水平的要求和生物医学工程领域科学技术的不断提高,在传统用于充当物理屏障,保持伤口愈合环境清洁的敷料类产品基础上,敷料类产品正逐渐向材料创新、功能多元和形式多样的方向发展。从我国最早于2002年发布的《医疗器械分类目录》[1](以下简称,“02 版《目录》”)和2017 年发布的新版《医疗器械分类目录》[2](以下简称,“新版《目录》”)的对比也可以看出,敷料的种类也从7 种增至22 种,新增了水胶体敷料、纤维敷料、碳纤维和活性炭敷料、含壳聚糖敷料、含银敷料、胶原贴敷料等新型敷料,其管理类别判定的依据更加细化。可即便如此,对于拟上市的敷料类产品的分类界定工作依然存在争议和挑战[3]。同时,2021 年新《条例》(国务院令第739 号)、《医疗器械注册与备案管理办法》及一系列配套法规文件的实施,意味着对于已上市的敷料类产品的监管也日趋严格,多地监管机构已对于敷料市场的乱象开展相关整治工作[4]。
对于审评机构而言,敷料类产品在延续注册过程中管理类别的判定则需要严格把关,避免出现“高类低延”的问题。根据广东省审评认证系统的不完全统计数据,新版《目录》于2018年8月1日实施后受理的第二类敷料类产品的延续注册申请事项有63 份左右(时间截至2022 年1 月1 日),其中有19 份左右涉及分类问题的发补,接近总申报量的三分之一。涉及分类发补的项目中有2份出具了不予批准的审评结论,3 份由申请人主动撤销,4 份要求注册人在注册证有效期届满6个月前按明确后的管理类别向相应药品监督管理部门申请注册,其他的则需要注册申请人对适用范围或结构组成等问题作出相应的规范或提供相关证据后才予以批准。可见,相对于其他的产品类别而言,敷料类产品在延续过程中管理类别不明确问题仍较为突出,这会导致注册周期的延长,甚至影响注册证的有效延续。以下将针对此类常见问题进行分析和探讨,为注册申请人和技术审评部门提供参考。
1.敷料类产品分类概述
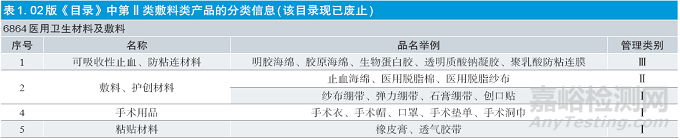
在新版《目录》实施以前,第二类敷料类产品在02 版《目录》中属“6864 医用卫生材料及敷料”的2——敷料、护创材料,如表1 所示。可以看出,敷料、护创类产品根据不同的品名或预期用途,仅按照Ⅰ类或Ⅱ类医疗器械进行管理(可吸收性止血、防粘连材料产品因其始终按Ⅲ类医疗器械管理,管理类别明确,故不在本文中讨论)。
此时,我国主要通过02 版《目录》结合对新产品进行分类界定的方式协同管理。随后,为了进一步满足医疗器械分类工作实践的需要,在2015 年7 月14 日,国家药品监督管理总局发布《医疗器械分类规则》[5](国家食品药品监督管理总局第15 号,以下简称,“规则”),该规则自2016 年1 月1 日起实施。规则中第七条内容:“医用敷料如果有以下情形,按照第三类医疗器械管理,包括:预期具有防组织或器官粘连功能,作为人工皮肤,接触真皮深层或其以下组织受损的创面,用于慢性创面,或者可被人体全部或部分吸收的”,首次明确了敷料类产品的管理类别涉及判定为第三类的主要原则。与此同时,其附件《医疗器械分类判定表》(见表2)还对如何根据敷料类产品的产品使用特征判定管理类别作了进一步细化。
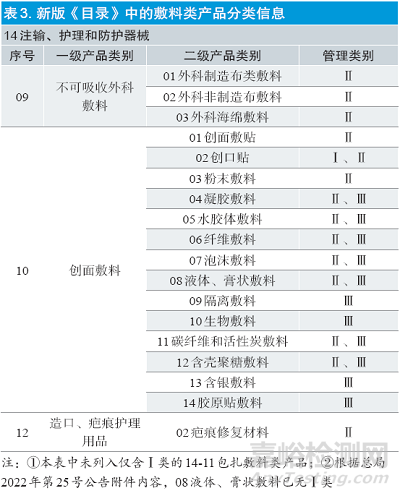
直至新版《目录》的发布,通过类别的分级管理,敷料产品的种类和管理类别变得更为具体(见表3)。原6864敷料类产品对应新版《目录》中的14-09 不可吸收外科敷料、14-10 创面敷料和14-11 包扎敷料(管理类别为Ⅰ类,本文不讨论)。此外,新增的12-02 疤痕修复材料也属于本文中讨论敷料的范畴。
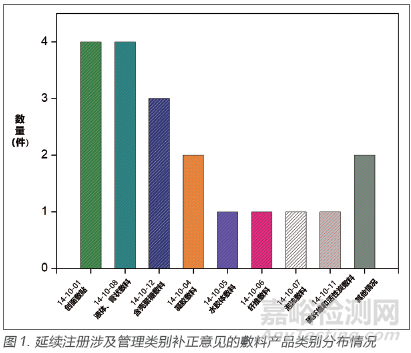
可以看出,相对于02 版《目录》而言,除可吸收性止血、防粘连材料的管理类别没有调整,仍作为Ⅲ类医疗器械进行管理外,其余的敷料类产品,从仅有Ⅰ类和Ⅱ类的2 个管理类别,衍生出根据不同的产品特性划分的更多更复杂的管理类别情况,部分新增的新型敷料还涉及Ⅲ类。反映在市场上,会出现即便产品名称相同,但产品的管理类别不同的现象。例如,国家食品药品监督管理总局曾在2010 年对一“泡沫敷料”产品作出分类界定通知(国食药监械[2010]111号[6]),明确将该类产品作为Ⅲ类医疗器械管理。但在新版《目录》实施后,原按此分类界定通知以Ⅲ类注册的产品如果符合新版《目录》中14-10-07 管理类别为Ⅱ类的情形,则可以按照Ⅱ类医疗器械申请延续注册。同理,应该会有较多的敷料类产品在新规实施后会涉及此类问题。仍以广东省的数据为例(图1),延续过程中涉及管理类别发补的问题主要集中在10-01 创面敷贴、10-08 液体和膏状敷料和10-12 含壳聚糖敷料上,其他类别则申报量较少,但也均有涉及,分布也相对平均。还有其他一些特殊情况的,如申请人宣称产品的分类属18-01-13 妇产科手术器械- 凝胶,但产品名称包含“敷料”,此类产品在延续时审评均提出了管理类别的补正意见。
2.延续注册过程中的常见问题
依据《广东省第二类医疗器械注册证延续办事指南》受理条件的要求:(一)原医疗器械注册证为广东省药品监督管理局核发,申报延续注册的产品依据《医疗器械分类目录》或分类界定通知等文件,管理类别属于二类医疗器械;(二)申请人是在广东省辖区范围依法进行登记的生产企业,原注册证为国家总局核发的三类医疗器械证,延续时产品类别依据《医疗器械分类目录》或分类界定通知等文件调整为二类医疗器械。此处需要强调的要求是,无论是原为Ⅱ类医疗器的产品,还是原为Ⅲ类调整为Ⅱ类的产品,均需要在延续注册申请的时候提交产品仍可以断定为Ⅱ类的依据,特别是对于分类不清或存在争议的产品。
能够作为分类依据的文件有《医疗器器械分类目》及其动态调整表、分类界定结果汇总文件、《免于临床评价医疗器械目录》以及其他总局发布有关某一特定类别产品的分类界定通告文件等。如果申请人未能提供前述文件作为明确分类依据的,将会涉及管理类别的补正意见。
2.1 未明确产品的二级产品类别
申请人在申请表或提交自行判断的分类说明文件仅说明了产品所属的子目录或一级产品类别,未列明产品所属的二级类别并提供完整的分类编码,故无法判定其在新版《目录》中的具体位置,以判定管理类别。此时,作为主体责任人,申请人应当对产品的属性和管理类别先进行自我判断并在申请延续注册时进行明确,审评机构才能够对申请人所提供分类依据的适宜性进行评估。
2.2 产品名称超范围表述或不规范
申报延续的产品名称中包含一些超出目录范围或不规范表述,而这些表述会影响到产品管理类别的判定。例如名称中“功能性”或“止血”等词语,“功能性”表述意味着产品的功能不具体、不明确,可以是某种功能也可以是某几种功能,而具有“止血”功能的敷料类产品则还需要进一步通过确认其所含成分是否会被人体吸收来判断其属于Ⅱ类还是Ⅲ类。
2.3 产品适用范围超范围表述或不规范
申请人虽明确了产品的分类编码,但前文中也有提到,部分敷料类产品在同一二级类别下有不同的管理类别。因此,这类产品当其适用范围和结构组成与新版《目录》或分类界定文件中差异较大,且未能明确能否用于慢性创面等涉及第三类产品界定的问题时,通常会涉及发补。例如适用范围中出现“各种创面”“溃疡”和“烧烫伤”等词语,“各种创面”属于夸大和超范围表述,皮肤性“溃疡”一般属于慢性创面,而“烧烫伤”也因其具有不同的分级需要分级限定。
2.4 产品结构组成涉及可吸收成分或药物
申报产品的结构组成超出新《目录》中产品描述中列出的材料或成分,根据目前的水平认知无法确认其是否会被人体吸收或发挥药理学作用时,申请人需提供相应的分类依据或支持的资料。如发挥药理学作用机制的,则应按相关要求做好分类界定工作,按药械组合产品申报或不作为医疗器械管理。
2.5 分类界定通知文件与产品信息不符
该问题分为两种情形,第一是在申报资料阶段,申请人提交申报产品以往的分类界定通知文件,但文中的产品名称、预期用途或产品描述等信息与申报产品注册证中载明内容差异较大,申请人无法提供合理解释说明,且经审评判断仍存在管理类别不明确问题的,仍会对产品的分类作出补正意见。第二是在补充资料阶段,申请人将补正期间取得的分类界定告知文件作为补充资料,此时,如存在产品描述信息差异的,也应提供充分合理的解释依据。通常来讲,其内容应保持一致,对于无法提供合理解释的,仍属产品管理类别不明确。
2.6 涉及降类产品延续的问题
已取得Ⅲ类注册证的产品在向原审批部门申请延续注册过程中,原审批部门对申报产品的管理类别作出补正意见,如建议申请人按Ⅱ类医疗器械向所在省市的药品监督管理部门申请延续。此时,建议申请人提交申报前与监管机构的既往沟通记录,即原审批部门对于申报产品申请延续注册时作出的有关分类的补正意见以及申请人的沟通回复(如适用)等作为按Ⅱ类申请延续时分类依据的支持性资料,以利于技术审评机构综合作出判断。
注册人依据现行法规自行判断申报产品属于管理类别由Ⅲ类调整为Ⅱ类,没有向原审批部门申请延续注册,直接向相应的食品药品监督管理部门申请注册延续。如果未能提交充分的分类依据,准确的分类编码信息、或所提供的分类信息与产品实际特征不符等,当技术审评人员依据申请人所提供现有申报产品资料判定其管理类别调整为第二类仍不充分时,同样会对申报产品的管理类别作出补正意见。
3.讨论与思考
依据《总局关于实施<医疗器械分类目录>有关事项的通告(2017 年第143号)》[7] 中的要求,自2018 年8月1 日起,注册人应当按照《医疗器械注册管理办法》(原总局令第4号,现为《医疗器械注册与备案管理办法》)和新版《目录》提出延续注册申请,即注册人应当根据申报产品的特征结合新版《目录》主动识别是否涉及管理类别调整的情形,并作出相应的注册计划。同时,通告中第七条提到涉及产品管理类别由高类别调整为低类别的,注册人应当在医疗器械注册证有效期届满6 个月前,按照改变后的类别向相应食品药品监督管理部门申请延续注册或者办理备案;涉及产品管理类别由低类别调整为高类别的,注册人应当按照改变后的类别向相应食品药品监督管理部门申请注册。由此可见:①对于已获证的Ⅱ类敷料类产品,如根据产品特征可明确产品的管理类别已调整为Ⅲ类,则申请人应自觉向国家药品监督管理局申请注册。存在争议的,建议注册人申请产品的分类界定,依据分类界定的结果进行注册申报;②对于已获证的Ⅲ类敷料产品,如根据产品特征可明确产品的管理类别已调整为Ⅱ类,则申请人可向省、自治区、直辖市药品监督管理部门申请Ⅱ类医疗器械延续注册审批事项。如存在争议的,仍建议注册人申请产品的分类界定,以作为按Ⅱ类医疗器械延续的分类依据。
此外,笔者在审评一些管理类别由Ⅲ类调整为Ⅱ类的案例时发现,由于此类产品的注册证原批准部门与改变类别后的审批部门不同,即申报系统不同,故当审评人员欲了解申报产品的首次核发或历次注册申报的具体信息时,仅能依赖于申请人所提供的申报材料,从而缺少了一些可以依据原产品的注册信息而判断产品管理类别的其他依据,进而可能会造成一些不必要的分类发补。由此引发思考,是否可以开发建立一种适用于类别转换产品的注册申请平台或申报资料共享平台,使这类产品的注册申报资料具有一定的连续性,从而在原注册审批部门及现审批部门可以实现同步,提高审评效率的同时也能够缩短申请人的注册周期。