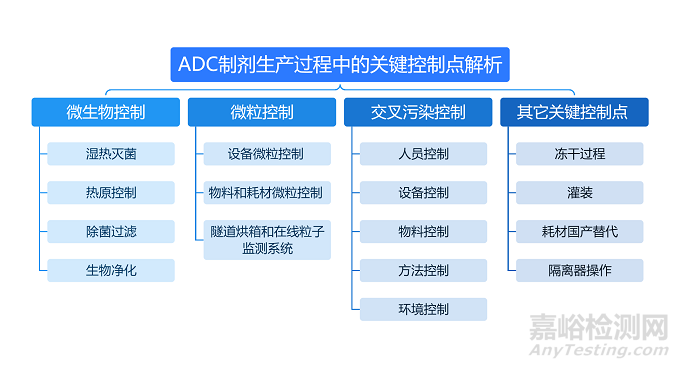
抗体偶联药物(ADC)兼具抗体的高特异性和细胞毒素的高抗肿瘤活性,对制剂生产也提出了更高要求。在ADC制剂生产中的经验,ADC制剂生产过程中的关键控制点,从微生物控制、微粒控制、交叉污染控制以及其它关键点控制。
本次讲详细解析ADC制剂生产过程中的“微生物控制”。
ADC制剂生产过程中的微生物控制
ADC制剂的生产和其他大分子的生产有很多相通之处,比如对微生物的控制。微生物控制主要包含湿热灭菌、热原控制、除菌过滤、生物净化四个方面:
一、湿热灭菌
湿热灭菌主要针对灭菌柜做器具灭菌,其中有五个注意点:
(一)灭菌柜性能:CD开发是确定灭菌柜性能的重要手段。平衡时间、BD测试和泄露率测试,这些性能测试也是考量灭菌柜质量的重要因素。
(二)针对灭菌柜的验证:包括空载热分布、装载热穿透、BI挑战以及最大装载/最小装载。灭菌柜在所有验证项目达到合格标准后才能投入生产使用。
(三)装载策略设计:这一部分考量的是装载的代表性以及涵盖范围。如果装载发生变化,应当在前期验证过程中尽可能地考量到,仅通过一个评估就能将变化涵盖,从而减少验证的工作量。装载发生变化后,需要重新验证的时间节点,也是设计装载时需要重点考量的部分。
(四)生产实际过程中的运行参数:验证通常采用Worse case参数,所以生产过程中会采用更加保守的策略。例如:验证时,灭菌条件为121℃、30min,而在实际生产中,可以考虑用122℃、30min的参数进行生产。这样可以保证在生产过程中,灭菌柜的温度即使低于122度,但只要高于121度,验证依然有效,可使风险降到最低。
(五)考虑Holding Time:通过相应的验证来确定灭菌后的存放时限。例如一些器具灭菌以后存放多长时间。该时限的确定通常有两个策略:一是通过相应的时限验证来确定,二是通过模拟灌装挑战来确定。
湿热灭菌的第二部分主要针对冻干机的SIP。
首先,CIP站作为为冻干机清洗提供介质的装置,需要考量是否进行相应的SIP。纯化水罐可以不进行灭菌,WFI罐则需要灭菌或至少进行蒸汽消毒处理,目的是控制微生物的限度。此外,冻干机SIP时需保证灭菌温度和时间,以符合灭菌的要求。
其次,冻干机的SIP过程一般采用过度杀灭。由于CIP结束后,冻干机并非处于干燥状态,导致微生物滋长,之后再进行SIP会有失败的风险,所以CIP后不要放置太长的时间,应尽快进行SIP。在SIP中,也要关注冻干机的通气过滤器。
第三是SIP的验证。该验证一般通过风险评估来确定探头的数量以及BI的数量和点位,如对于冻干箱箱体的板层、内壁和大门,就需要通过风险评估来考虑在哪些地方布置探头和BI。SIP的管路和过滤器则必须布置探头和BI,以此确保管路和过滤器能够达到无菌效果。BI在布置的过程中一定要牢固,且不能堵塞管路。如果布置不合适,可能导致SIP结束后,BI被蒸汽冲走,产生引入污染的风险,或者堵塞管路,最终造成SIP的失败。
最后是SIP的时效性。与灭菌器柜相同,需要考察其Holding Time。该考察可以通过验证来确定,也可以进行三批模拟灌装来确定灭菌的时效性。通气过滤器的灭菌次数也很关键,要根据过滤器的耐受次数以及实际的生产情况来综合考察。
二、热原控制
微生物控制第二部分是热原控制,主要涉及隧道烘箱,可以从五个方面来解析:
(一)隧道烘箱的运行模式。在生产过程中采用的生产模式通常叫日间模式;在生产停止过程中,恰当的方式是切换成夜间模式,保持隧道烘箱的送风,防止因送风关闭造成隧道烘箱污染。
(二)冷却段灭菌。一般每批生产前需要对冷却段进行灭菌,保证冷却段的无菌环境。
(三)冷却段压差梯度的设计。特别是对于ADC的生产,一般会使用隔离器来保证活性药物生产的安全性,所以隧道烘箱的出口段压差比普通隧道烘箱高。这对隧道烘箱的挑战较大,需要防止隔离器的风灌到隧道烘箱,还要防止冷却段的风灌到加热段。采用双级的压差梯度可以避免冷却段对高温段的影响。
(四)隧道烘箱本身各段的压差。要与设计保持一致。在实际运行中不能偏离设计,否则可能造成倒灌,进而影响除热原的效果。
(五)温度和网带速度。这个时候要考察不同规格的西林瓶对应的除热原温度、网带速度存在差别,这些参数需要通过验证来确定。
三、除菌过滤
微生物控制需要考虑的第三部分是除菌过滤,主要是药液的除菌过滤。
具体包含以下六个方面:
(一)滤膜材质和膜面积的选择。目前在市面上,滤膜材质多用PVDF和PES。在实际操作中,材质的选择要根据药液的特性确定,通常是根据药液的兼容性选择。膜面积的选择要留有缓冲余地,如果仅通过计算的值来选择,没有留有缓冲,可能会造成滤器的堵塞和破损,尤其要注意高粘度、高浓度、有蛋白聚集的情况,可以考虑采用预过滤和多级滤器并联的方式来有效降低风险。
(二)时限研究。混匀后至过滤前一定要考察时限,还要考虑药液整体的过滤时间、药液过滤后的存放时间。
(三)影响药液除菌过滤效能的因素。其中最主要的是药液本身的性质,此外还有过滤工艺参数,如过滤的压力、以及过滤器和药液相互的作用,还有过滤量和使用周期。
(四)无菌连接。无菌连接器市面上也有很多种。此时要重点考察厂家对无菌连接器是否进行了相应验证、验证资料是否齐全、整体的密封性以及无菌连接器辐照灭菌验证资料是否齐备。因为过滤的时候要考量滤器前后端的压差,所以也要有相应的压力监测系统,还要有相应的记录及进行相应的计量。
(五)一次性系统。根据EU GMP规范,使用前要进行完整性测试。这时候要考量的是在无菌状态下,做完整性测试过程中的通气、通水经过相应的过滤,应当怎样收集过滤的水。
(六)微生物负荷。需要重点考察的是取样量。法规要求是10CFU / 100ml。ADC和其他大分子药物附加值比较高,通常会采用折中的方案。例如取3ml,规定定量的CFU。微生物负荷的取样时间,也是一个考量点。一般的取样会在除菌过滤前。如果除菌过滤是持续的过程,那就要在除菌过滤前的最后时间点进行取样。
四、生物净化
第四部分就是ADC制剂生产过程中微生物控制的非常重要的一部分,即生物净化,通常指隔离器VHP。
隔离器VHP需要重点关注四个方面:
(一)CD/CV的开发。隔离器的温度、湿度、压差以及蒸发盘的温度、过氧化氢的注入量、持续时间,都是在CD/CV开发过程中重点考量的参数。
(二)BI的选择与处理。BI的选择重点考虑D值;BI的保存要注意温湿度以及避免裸手接触BI。验证过程可考虑分数法,每个点布置3个BI,防止出现个别阳性的情况。
(三)VHP实施过程中的控制。要关注几个点:一是VHP的起始阶段,即除湿后的湿度。还要关注VHP过程中(提升阶段)过氧化氢达到的浓度,该浓度需达到一定的数值,且经过验证。如验证的时候达到400PPM才能达到杀灭的效果,但如果实际过程中,浓度没有达到400PPM则会造成风险,这就不是成功的VHP过程。净化阶段持续时间也很重要,比如验证过程中进行的是85分钟的验证,生产过程中就会更保守,延长时间,真正对无菌保证起到双保险的作用。
(四)过氧化氢排残。一般要配备高低浓度探头,有的设备商只配了一种探头,并宣称高低浓度均可监测。实际上这种探头对高浓度比较精确,而低浓度时误差非常大,所以并不适宜。比较适宜的做法是分开设置浓度探头,并且要确认低浓度探头的精度范围。很多时候,低浓度探头在1ppm的精度不高,排残的时候显示到了1ppm,如果以更精确的便携式监测的话,可能有几个ppm,这对于敏感性高的药品有一定影响。同时,低浓度探头的安装位置也很重要。
在排残时需要在验证过程中做实验,保证过氧化氢的浓度达到实际的数值,而不是在隔离器本身的探头里面显示的数值。因为探头精度有限,一些特殊药品对过氧化氢的敏感度非常高,此时需要单独考察,考察时要做到:一是延长排残时间,二是以相应的方法进行确认。