您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-03-11 22:33
美国FDA将医疗器械按照风险从低到高划分为三类,即Ⅰ类、Ⅱ类及Ⅲ类。不同管理类别的器械上市途径不同,部分I类、大多数II类及少数Ⅲ类器械需通过上市前通告510(k)申请获得市场准入许可,该途径占上市申请器械的60%左右。
第三方 510(k)审核程序是FDA为医疗器械注册申请人提供的一个自愿的替代性的审核程序,该程序允许有资质认可的第三方 510(k)审核机构(Third Party (3P510k) Review Organization)审核规定的中、低风险的医疗器械,以提高510(k)的审核效率。您家的医疗器械可以申请第三方510(k)审核吗?今天,小编即为您解答。
一、FDA第三方 510(k)审核基本流程
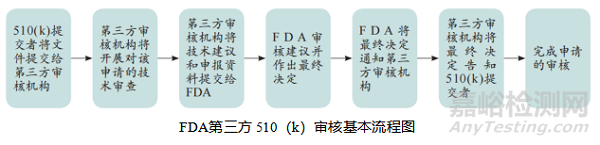
确定申报产品是否在第三方审核项目范围内
查询产品代码分类数据库或FDA第三方审核项目公开网站,以及是否具备相应专家,以判定申报产品类型是否属于第三方审核范围。当前第三方审核项目中的产品包括麻醉学、耳鼻喉、血液学、产科/妇科、心血管及临床化学等20类产品。FDA会动态更新产品列表,使FDA始终可将审查资源集中于高风险及复杂设备,同时保持对第三方审核项目的高度信任。
如不是,将拒绝该510(k)申请;如是,则为该510(k)申请分派合适的技术审评人员。若必要,将分派具有相关专业背景的技术专家。
申请材料完整性审查
申请人提交510(k)申请前,应将与FDA间的所有咨询交流信息告知机构。
机构根据21CFR 807.87至807.100中的法规规定,对递交资料进行受理审查,以确定该申请是否包含所有必要信息,并能得出实质性等同(SE)或非实质性等同(NSE)的决定。FDA推荐机构使用拒绝接受清单[Refuse to accept (RTA) checklist]以判定申报资料是否完整。
实质性等同评审
机构充分利用FDA公开发布的有关信息,包括产品类指导原则、通用指导原则及联邦法规21章(Title 21-Code of Federal Regulations,21 CFR)中对特定II类产品的特殊控制要求。机构应查阅上市后数据库,了解临床使用有关问题或风险。FDA鼓励机构及时与FDA有关审评人员进行沟通咨询,特别是审评人员首次审评某一类产品时。
实质性等同的具体判定路径可参考相应指导原则。机构应在内部指定一位审定员,其不参与此前的审评工作并负责对审评小组的审评建议提出最终评估结论。该审定员应具有充分的权威和能力,确保可独立评估审评建议的质量及可接受度。
如何处理510(k)申报资料中的缺陷
技术审评过程中,如机构认为申报资料存在缺陷或问题,可采用电话及邮件等方式与申请人沟通。所有沟通交流记录应规范移交至FDA,同时确保机构不能变成申请人的咨询机构。
出具审评建议
一旦机构完成审查,应出具一份审评建议,该建议文档应说明理由并作出上述审评建议的具体步骤,有助于避免FDA对510(k)申请资料重新审查,提高审评效率。
机构将有关资料移交FDA
完成审评后,机构中的审定员递交510(k)申报资料及审评建议书的电子文档至FDA文档控制中心。FDA须在收到材料后的30日内做出最终决定。若申报资料存在缺陷,FDA将具体问题告知机构,并由后者通知申请人补充信息。
二、FDA对第三方510(k)审核机构的监管
符合第三方审核项目的产品类型及考量因素
FDA在2020年发布的第三方510(k)审核项目指南中给出了在确定产品类型是否符合第三方审核项目的考虑因素,主要内容如下:
(1) 产品风险。III类设备不符合第三方审核资格,若产品类型为永久植入、维持生命或生命支持,除非理由充足,否则不会被纳入第三方审核项目。
(2) 产品类型的难易程度。如具有新技术的产品,包括某些通过De Novo程序初步分类的需要复杂特殊控制的产品,可能不符合第三方审核条件。
(3) 获取充分信息的程度。如果与评估该产品类型的相关信息不能在机构外共享(如其为FDA内部专有),则该产品类型可能不符合第三方审核资格。
(4) 上市后数据表明该产品类型本身存在安全风险。如某产品上市后存在安全问题,FDA可能将该产品从第三方审核项目产品清单中移除。
机构的资质管理
FDA对机构的资质认定将在授予认定之日起3年内有效。FDA建议机构在其资质认定状态到期前至少60日申请重新资质认定。FDA会定期(至少每3年1次)或因地对每个机构的运行情况及所开展的审核项目进行评估。如评估结果显示该机构不符合FD&C Act法第523条,FDA可采取措施暂停或撤销第三方审核资质。
三、第三方 510(k)审核常用网址
当前符合第三方审核条件的器械清单
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfThirdParty/current.cfm
FDA认可的第三方 510(k)审核机构清单
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfthirdparty/accredit.cfm?party_key=8
申请材料拒绝接受清单 [Refuse to accept (RTA) checklist]
https://www.fda.gov/media/83888/download
参考资料:俞卉,袁鹏,杨挺,朱文武.关于美国FDA第三方510(k)审核项目的分析研究[J].中国医学装备,2021,18(05):191-195.

来源:苏大卫环


