您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-10-14 23:40
A:Q6A主要介绍新原料药和新药制剂的质量标准,它为化学合成的新原料药及其制剂检测方法的选择、检测限度的制定和论证提供了指导。在Q6A中如何定义质量标准?
B:质量标准由一系列的检测项目、分析方法和认可限度组成,英文中对应的单词为specification。产品要符合标准是指原料药和制剂按照给定的方法检测,其结果应符合认可限度。质量标准是非常重要的质量指标,必须经过管理机构的批准并且还要作为产品注册的批准依据。
质量标准是确保原料药和制剂的质量及一致性质量控制体系的一部分。质量控制体系其他部分还包括制订质量标准所依据的研发期间获得的全部产品性质和GMP执行情况,如设施、已验证的生产工艺和检测方法、原材料的检验、生产过程中的检验以及稳定性试验等。
质量标准是用来进一步确认原料药和制剂的质量,而不是体现产品的所有性质,故在质量标准中应重点设定能反映药物安全性、有效性的检测项目。
A:Q6A指导原则适用的范围是什么?
B:本指导原则只适用于申请上市的新药制剂(包括复方制剂)或新原料药,不涵盖临床研究阶段的药物。适用剂型包括口服固体制剂、口服液体制剂和非肠道给药制剂(大、小容量),但这并不限制该指导原则对其他剂型的适用性。本指导原则中所提及的剂型可作为例子,这些例子可用于其他未讨论到的剂型。对于其他剂型如吸入剂(粉末状、溶液等)、局部用药制剂(乳膏、软膏、凝胶剂)和透皮制剂,也鼓励应用本指导原则中的概念。
本指导原则不适用于高分子肽、多肽、生物制品。不涵盖放射性药物、发酵制品、寡聚核苷酸、草药和来源于动植物的粗制品。
A:针对本指导原则有哪些背景知识需要了解?
B:第一个概念是货架期标准与放行标准。对于药物制剂,放行与货架期标准限度可以不同,通常放行标准限度严格于货架期标准限度,尤其是对含量和杂质的限度要求。在日本和美国,这一组概念只用于内控标准,而不用在法定标准中。因此在这些地区,法定的认可标准从放行出厂到货架寿命结束期间均是相同的。但申报者可选用更严格的内控标准作为放行依据,以确保产品在货架寿命期间仍符合法定的认可标准。在欧盟,当放行和货架期标准不同时,管理机构要求提供各自的标准。
第二个概念是参数放行。在某些情况下,比如最终灭菌制剂的无菌检验,可以通过经批准的参数放行方法替代常规的放行检验。在此情况下,每个批次的放行取决于对特定参数监测结果的满意度,即对制剂生产最终灭菌阶段的温度、压力和时间。这些参数通常可以被更精确地控制和测定,因此在判断无菌结果时,它们比最终成品的无菌检测结果更可靠,但前提是灭菌工艺已经过了充分的验证。在进行参数放行时,仍应在质量标准中制订未直接控制的项目及与其相关的检测方法。
第三个背景知识是关于药典的统一化,Q6指导原则的充分实施,取决于是否能与质量标准中涉及一些常规项目的药典分析方法相互协调。一旦取得协调,就可以在三个地区的质量标准中采用统一的方法和认可限度。
A:在研发中如何确定某个质量标准?
B:最初提出质量标准时,应对每一个检测方法和每一个认可限度的合理性进行论证。论证应以新原料药的合成和制剂生产过程中获得的数据为基础,考虑的因素包括有关的研究开发数据、药典标准、用于毒理和临床研究的原料药及制剂的检测数据、加速试验和长期稳定性研究的结果;此外还应考虑分析方法和生产可能波动的合理范围。在制订检验项目和认可限度时,首先应考虑到稳定性批次和生产规模放大时验证批次的检测结果。若计划有多个生产场所,则在建立最初的检验项目和认可限度时,就应充分考虑所有生产场所所能够获得的数据。
A:对于原料药和制剂,需要在质量标准中列出哪些检测项目?
B:新原料药和新药制剂的检测项目分常规检测和专属检测两类。在常规检测中两者检测项目相同,包括性状、鉴别、含量测定和杂质。鉴别试验对原料药应具有专属性,如采用红外分光光度法。只将一个色谱保留时间作为鉴别依据并不具有专属性,但可以用两种不同原理的色谱方法或者是用一种色谱方法与其他试验相结合进行鉴别。若新原料药是盐,则应对每个离子进行专属鉴别试验,还有一个对盐本身的专属性试验。具有光学活性的新原料药,也需进行专属性鉴别或者手性含量测定。
对于含量测定,许多情况下可能会使用同样的方法(如高效液相色谱法)同时测定含量和杂质量。但若含量测定采用非专属性方法,应额外采用另一种方法来补充完善其专属性。如若采用滴定法测定含量,应同时选用适当的专属性方法测定杂质。
杂质包括有机杂质、无机杂质(降解产物)和残留溶剂。新原料药降解产生的有机杂质和该制剂在生产过程中产生的杂质均应在新药制剂中检测。应对单个特定降解产物(包括已鉴定的和未鉴定的)及总降解产物的可接受限度进行规定。要注意的是新原料药合成中生成的杂质(工艺杂质)通常在原料药的检测中已控制,因此不包括在制剂总杂质限度中。但当工艺杂质同时也是降解物时,应监测其含量并列入总降解产物的限度中。
A:杂质和降解产物有什么区别?
B:药物分子长时间放置和(或)受光、温度、pH值、水的作用或与辅料和(或)直接接触容器/密闭系统反应,发生化学变化而产生的分子称降解产物,也称为分解产物。杂质是指原料药中非原料药实体的任何成分以及制剂中活性成分实体或辅料以外的任何成分。
A:专属检测包括哪些检测项目?
B:专属检测中新原料药和制剂的检测项目并不相同。新原料药包括物理化学性质、粒径、多晶型、微生物限度、无机杂质、水分和新手性原料药检测。这些项目通常会影响原料药的质量,因此要在质量标准中规定检测限度要求。
物理化学性质包括水溶液的pH值、熔点/熔距、折光系数等,通常检测方法较为独特,如采用毛细管测熔点,阿贝折射仪测折光。这类检测项目的设立主要取决于新原料药的物理性质以及预期用途。
粒径检测对于要制成固体制剂或者混悬剂的新原料药而言十分重要,因为粒径大小可能会影响溶出速率、生物利用度或者稳定性。此外,还应考虑粒度大小是否为影响生产的关键因素,在这种情况下需要采用适当方法测定粒径以及粒径分布,并建立认可限度。
多晶型包括溶剂化物、水合物和无定型物。有些新原料药以不同晶型存在,不同的晶型物理性质不同。在一些情况下,晶型不同会影响新药制剂的质量或者功效。若已证明原料药存在不同晶型并且不同晶型会影响制剂的功效、生物利用度或者稳定性,则此时应将多晶型的含量测定作为检测项目,规定晶型的质量标准。
A:ICH还对其他检测项目提出了哪些要求?
B:对于微生物检测,需要规定需氧菌、酵母和霉菌的总数以及无法检测的特定致病菌,如金黄色葡萄球菌、铜绿假单胞菌。应根据经验和已有数据确定生产过程中的抽样频次或者时间点。应通过原料药性质、生产方式和制剂预期用途等因素确定微生物检测的种类和认可限度。如无菌原料药应设定无菌检测,用于注射剂的原料药应进行细菌内毒素检测。
关于杂质的相关问题已在Q3中进行了详细的说明。若新原料药易吸湿或吸湿后易降解,或者原料药含结晶水,则水分检测十分重要。可根据结晶水的多少或吸湿量的数据来确定认可限度。某些情况下,也可采用干燥失重测定水分,但应首选专属性好的测定方法,如卡尔费休法。
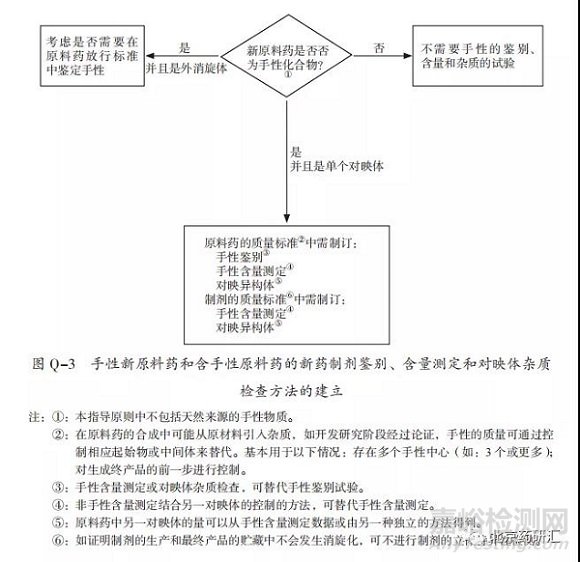
对于新手性原料药检测,图Q-3展示的是手性新原料药和含手性原料药的新药制剂的鉴别、含量测定和对映体杂质检查方法的建立过程。需要注意Q6A指导原则中这项内容的检测并不包括天然来源的手性物质。
A:新药制剂有哪些专属性检测项目?
B:新药制剂剂型可分为固体口服制剂、液体口服制剂和非肠道给药制剂。三类都需要做的检测项目是单位剂量均匀度,水分和微生物限度。单位剂量均匀度包括制剂的重量差异,制剂中活性成分的含量均匀度两种概念,全部采用药典方法测定。通常在质量标准中只列入其中之一,不同时包括两项检测。对于超过了允许用重量差异检验均匀度界限的制剂采用重量差异作检查时,申报者应该在药物开发阶段就证明制剂是足够均匀的。
A:口服固体制剂的质量标准中通常包括测定原料药从制剂中释放的试验,即溶出度试验。常释制剂和缓释制剂的溶出度试验内容有何不同?
B:对常释制剂而言,通常进行单点测定。如果已证明溶出行为会显著影响生物利用度,应建立能辨别生物利用度不好批次的溶出试验条件。如果处方和工艺的改变显著影响溶出度,而这些改变又不能用质量标准中的其他项目来控制,也应采用能区分这些变化的溶出试验条件。如果溶出度显著影响生物利用度,可接受标准应能剔除生物利用度不好的批次。换言之,应制订仅临床可接受批次能通过的试验条件和可接受标准。
对于缓释制剂,应采用多时间点取样。如果可获得不同释放速率处方的人体生物利用度数据,则可根据体内/体外相关性来设置可接受标准;如果没有这些数据,而且药物释放与体外试验条件显示依赖关系时,则应根据已获得的批次数据来制订可接受标准。
A:口服固体制剂需要进行硬度/脆碎度测试吗?
B:通常硬度/脆碎度检测作为过程检测,质量标准中不必包括这些项目。但如果硬度和脆碎度对制剂质量有重要影响(如咀嚼片),则应在质量标准中制订相应的可接受标准。
A:什么叫作过程检测?
B:过程检测是指在原料药或制剂生产过程中进行的检测,不属于出厂前的正式批检验。某些生产过程中的检测,仅仅是为了在某一操作范围内调节工艺参数,如包衣前片心的硬度、脆碎度及片重,质量标准中一般不包括这些检测项目。某些生产工艺中进行的检测项目,其可接受标准与放行标准要求一致或更严格(如溶液的pH值)。当某个检测项目纳入质量标准中时,即使能充分满足质量标准的要求,但依旧应对该方法进行验证,以表明检测结果或产品性能特性从生产阶段到成品未发生改变。
A:口服液体制剂中通常含有抑菌剂和抗氧剂,对这两项含量测定分别有哪些要求?
B:对需加入抑菌剂的口服液体制剂,应制订抑菌剂的含量测定及可接受标准。抑菌剂含量可接受标准应根据在整个使用期间和货架期能保证制剂微生物限度符合要求的抑菌剂水平确定。应采用药典中抑菌剂抗微生物有效性试验来确定抑菌剂的最低有效抑菌浓度。尽管质量标准中通常包括化学测定抑菌剂的含量,但在开发阶段、规模放大阶段和整个货架期(如在稳定性试验中,参照ICH的新原料药及制剂稳定性试验原则)都应证明抑菌剂的有效性。
通常放行时要进行抑菌剂和抗氧剂含量测定。在某些情况下,如有开发和稳定性数据的支持,货架期标准中可不再制订抑菌剂和抗氧剂的含量测定,过程检测可替代放行检测。若生产过程中进行了抑菌剂和抗氧剂的含量测定,应仍将其可接受标准制订在质量标准中。若仅进行放行检验,则无论是生产工艺还是容器/密闭系统发生改变,都应对此进行重新研究。
A:对于相对黏稠的溶液或者混悬液而言,都有哪些测试项目?
B:通常需要进行溶出试验,根据体内生物利用度良好的多批次样品的溶出曲线以及变异范围,确定可接受标准。还应建立粒度分布检测方法并制定可接受标准,可接受标准应包括粒度分布(即在一定粒径范围内的粒子数占粒子总数的百分比),并应严格规定粒径的平均值、上限和(或)下限。如果在开发阶段已经证明这些产品始终保持快速释放,则质量标准中可不列入粒度分布测定。在决定采用溶出度测定还是粒度分布测定时,应充分考虑开发阶段获得的数据。经过充分论证,粒度分布测定可代替溶出度测定。
此外还有流体学特性测试和再分散性测试,根据产品开发阶段所积累的数据,提出进行定期抽检还是从质量标准中略去此项。
A:对于非肠道给药制剂,有哪些需要注意的地方吗?
B:所有的非肠道给药制剂都应进行无菌检查,制订检测方法和可接受标准。如果在开发和验证阶段所积累的数据证明参数放行是可行的,可将参数放行应用于终端灭菌制剂。此外,非肠道给药制剂必须制订不溶性微粒的可接受标准,通常包括可见微粒和(或)溶液澄清度以及在显微镜下可见的微粒。
对于包装在预填充注射器、自动注射盒或相当的容器中的非肠道给药制剂,应制订与给药系统功能性相关的检测项目和可接受标准。包括可注射性、压力、密封性(泄漏)和(或)一些参数,如滴帽移动力、活塞释放力、活塞移动力、动力注射器作用力。在某些情况下,这些项目可以在生产过程中进行检测。根据产品开发期间所积累的数据,可以提出进行跳检,或是从质量标准中剔除部分或全部测试项目。

来源:Internet


