您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2019-11-12 15:55
经历了这一段时间的大浪淘沙之后, 行业内对制剂开发的思考也有了明显的改变,本文系从药物在体内的吸收过程的理解来论述其对制剂开发的影响。
前言
本文主要阐述经消化道吸收的固体口服制剂的体内过程,通过其过程的了解来认识行业在制剂开发中所进行的探索。众所周知,固体制剂要实现体内吸收必须经历三个阶段,其一药物颗粒在体内溶出环境的暴露;其二,药物在胃肠环境溶解成分子或离子形态;其三,溶解后药物分子经生物膜渗透入血进而运输到局部或者全身进行疾病治疗。从笔者上述描述可以发现其中最核心的内容就是理解药物所处的“环境”问题,只有仔细分析体内环境问题,我们才能更好的评估和选择体外环境。下文将从空间、时间以及化学环境三个维度简述药物体内吸收过程以及相关的思考。
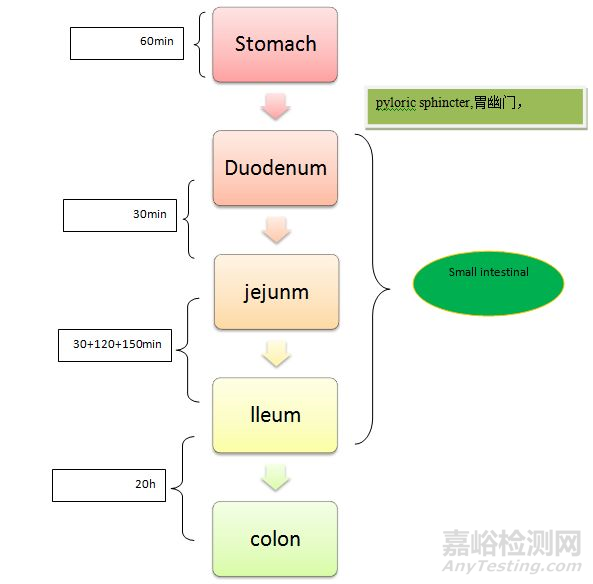
空间维度
如下图所示,固体制剂经温水服用后,通过食道经贲门后进入胃环境(食道内径约为2-3cm,亦有资料提及食道内径约为17-23mm,扩张条件下可达35mm);胃是口服药物进入胃肠道的主要通道 ,在这里速释(IR)制剂快速崩解,使活性药物成分(原料药)能够在它到达吸收部位已实现完全溶解,通常情况下药物经一定水量送服之后胃容积约达到300-500ml;药物抵达胃部之后会经胃排空作用使之从为幽门(Pylorus)进入十二指肠(十二指肠顾名思义约为12个指头的并列长度,约为20+cm),十二指肠是物质从胃部向肠道过渡的主要部位与之相连的胰腺(pancreas)和胆囊(gallbladder)对其环境的改变有重要影响;经十二指肠后进入最主要的吸收部位——空肠以及回肠,人体小肠由于其特有的褶皱以及微绒毛等结构使之具有约200m2 吸收表面积,如此巨大的表面特性为药物吸收提供了广阔的环境, 一般情况下药物到达该部位已基本完全释放,在此部位主要是药物的跨生物膜转运阶段。
然而药物的跨膜转运模式多种多样,不同的转运模式所依赖的膜因素也是不同的,较为常见的模式为被动转运机制,该机制下在肠道内溶解的药物经扩散作用依次经微绒毛表面的“不流动水层”、上皮细胞的“磷脂双分子层”、上皮细胞内部以及跨红细胞等一系列分布分配作用最终入血,经血红蛋白结合或者以游离形式随血液循环抵达病灶部位。(上述即为药物分子扩散至不流动水层后经8层磷脂层及细胞质、组织液、亚细胞器等最终抵达血液的过程);通常情况下药物会在小肠段完成吸收,但对于缓释控释制剂而言,结肠亦可能是其吸收部位,当然结肠结构不同于小肠结构不具备广阔的吸收表面积,但其特殊的升降结构为缓控释的体内滞留提供了空间条件。
时间维度
药物经口腔服用后抵达胃部的时间是较快的,通常情况下10+s 即可抵达胃部,胃部药物体转运时间受食物影响显著,空腹状态下胃排空时间和餐后有显著的差异。一般餐后状态下胃排空的时间约为4h,胃排空受物质状态影响排除速度一般满足:液体>固体>粘稠状物质;药物经幽门输出后,在小肠系统的驻留时间一般在3.5h-4.5h;结肠内物质转运速度较慢,对于药物而言升结肠处由于依然具备药物溶解的水性环境故而还可以考虑在内,此处药物最长还可以获得约24h的滞留1。
化学环境
药物体内的化学环境的变化是制剂开发者普遍关注的内容,众所周知,在禁食条件下一个健康的人胃是酸性的,范围在1到3之间,故而对于难溶性弱碱该pH环境可为之提供最有利的溶解环境,对于胃酸分泌不足的患者或老年人而言胃内pH值会升到pH5.0附近。然当进入十二指肠之后,与之相连的胰腺系统以及胆囊系统受幽门排除物低pH的影响,为避免胃内低pH对肠壁细胞产生破坏胰腺分泌高浓度的碳酸氢根离子对酸性介质进行中和使其pH上升到5~6左右,胆囊则分泌胆汁对食物的糜化以及提高难溶性药物的溶解度;结肠部位是水分以及电解质主要吸收的部位,营养物质以及药物分子大多经小肠吸收,然由于升结肠部位任然可以提供药物溶解的水环境故而此部位依然可以作为部分控释制剂设计的考虑方向。
体内过程指导产品开发
基于药物体内吸收的理解,可以更好的理解产品设计过程中相关要求或者标准的制定依据。也可以根据人体本身的生理状况来设计制剂产品,例如基于胃环境的ph理解,在开发胃内不稳定产品时选择胃不溶但能肠溶的包衣材料来保护药物有效成分;亦可以根据胃环境对不同物理尺寸或状态的固体的排空时间来评估不同剂型以及不同处方的体内吸收差异;也可根据产品体内吸收数据以及化合物溶解渗透等属性来评估体外释放需求等。
于此同时,许多指导原则以及评价方法制定都是基于对体内吸收过程的理解之上建立起来的。例如产品设计中物理尺寸的限定、BCS分类中判断环境的选择、速释制剂豁免的溶出要求、甚至制剂产品的溶出方法选择、产品质量的体外评价要求以及漏槽条件的要求等等。
体内过程指导溶出方法开发
溶出方法的开发对仿制药开发具有非常重要的影响,建立具有体内预测力的溶出方法系该领域最高的追求。目前常用的溶出方法主要是基于药物体内历经的pH环境的理解建立起来的,该类介质的选择科学性即是这一系列pH缓冲液从pH的维度模拟了药物可能遇到的环境,而局限性也在于仅仅只能模拟pH这一维度而并不具备体内生物相关性。
基于此1998年Dressman,J.B等人发布了禁食以及餐后的两种模拟体内小肠环境的溶出介质( FaSSIF以及FeSSIF),在结合更多色体内外实验数据对我基础上Vertzoni等人对上述两种介质进行了进一步的优化并加入了人体胃肠环境中的各项基础物质并将肠内模拟介质推广到胃环境的空腹与餐后两种状态2。
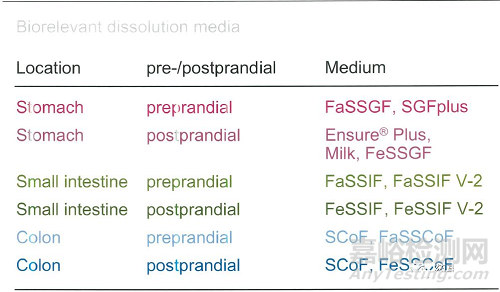
为了成功预测体内药物行为,溶解介质组合情况是需要考虑的,当然介质的体积和流体动力学等特性也应考量。故而在溶出介质体积以及装置选择方面最好根据生理状况来调整,测试仪器的实际约束条件也需要得到满足。根据一些关于胃肠液量的报告,流体体积在最初禁食时胃容量约为300ml在进食的状态下该体积则会增加,而在小肠中禁食时体积为200ml餐后体积约为1L(药物与肠壁不可能整体都接触到故而在考察以肠道为主要溶出环境的制剂产品时不宜选择如此大的体积)。
装置方面则应该结合药物的特性进行装置的适当选择。基于体内过程的了解选择生物类似性溶出介质和适当装置的建立具有预测力的溶出方法。 Ekarat Jantratid3等人以双氯芬酸钠缓释微丸为模型药物对比了USP 的QC方法和生物类似介质下往复桶法(USP 第三法)以及生物类似介质下流通池法(USP 第四法)中的溶出趋势,如下图
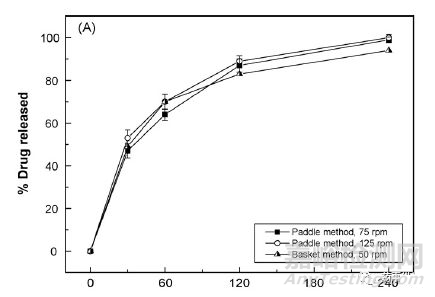
Figure 1 QC 溶出条件下溶出曲线
上图显示在QC介质以及不同转速以及不同装置条件下溶出趋势无显著差异,并且观察到缓释制剂在120min内即可完成85%以上的释放。
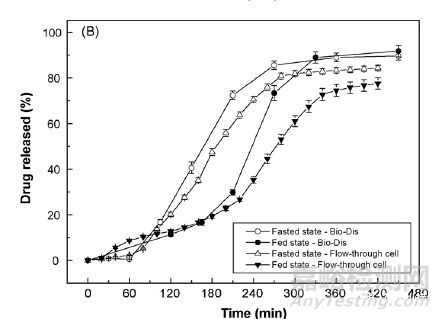
Figure 2 生物类似介质往复桶法和流通池法中溶出曲线
上图显示在模拟空腹和餐后的溶出介质中,往复桶法和流通池法均表现出相似的溶出趋势即模拟空腹介质中溶出均快于模拟餐后介质。该信息提示双氯芬酸钠缓释微丸在体内不同进食条件下或将表现出不同的释放趋势,然QC方法是无法给予这方面信息的支持。基于上述假设进行了模型药物的体内研究。
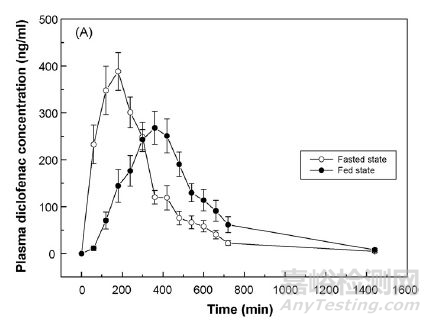
Figure 3 空腹以及餐后体内吸收曲线
上图表明,双氯芬酸钠缓释微丸在空腹状态下体内达峰时间要早于餐后,这一结果与体外溶出方法中释放速度的差异是一致的。基于双氯芬酸钠在体内消除速率信息结合PK数据进行翻卷积研究最终获得模拟体内介质+往复桶法和模拟体内介质+流通池法条件下IVIVC特征。
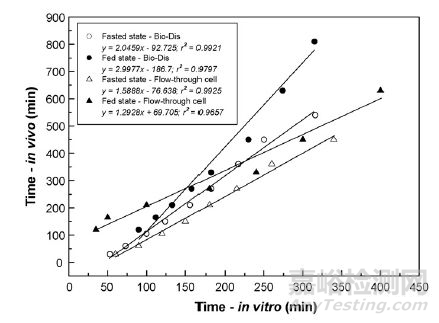
Figure 4 往复桶法与流通池法的IVIVC
上述案例说明利用模拟体内介质+往复桶法以及流通池方法进行双氯芬酸钠缓释微丸的溶出考察时可实现较好的IVIVC方法,然对于这种化合物用桨法或者篮法来模拟体内过程都是很难实现的。值得注意的是:预测力方法的开发永远是case by case,因为该方法的建立不仅受介质开发和装置开发的限制还与药物本身的性质息息相关。
对每一个产品开发一套预测力方法是不可能实现的,对于缓释(MR)制剂而言,由于该类制剂在体内经历的生理环境是不断变化的,故而使用一系列生物相关介质模拟药物胃通过小肠的变化甚至结肠的过程是有必要的。
基于此GastroPlus 软件应运而生,该软件系基于人体胃肠道生理特性、 以高级房室模型( ACAT 模型) 作吸收模型、生理药动学模型( PBPK) 作处置模型,模拟不同剂型的药物经多种途径给药后体内过程的理解建立的,该类方法在仿制药开发中以取得FDA等多国药监督部门的认可,该方法最大的优点在于在制剂开发过程中可以开发出具有体内预测力的溶出方法,基于此溶出方法可以更好的指导仿制药开发。
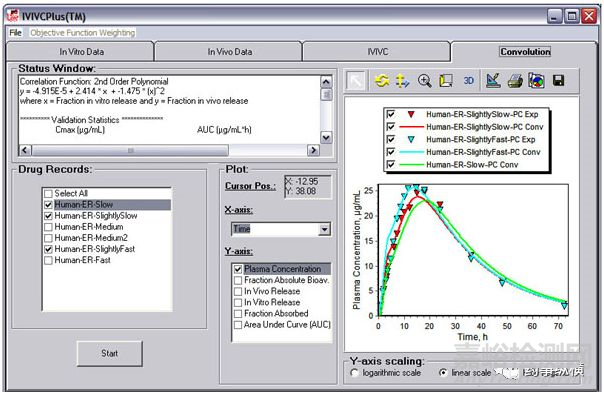
潘瑞雪4等采用不同溶出度测定方法考察考了国产β-内酰胺类抗生素在不同介质中的溶出行为,并通过GastroPlus模拟不同释放速度的制剂在体内的吸收情况,进而实现不同厂家制剂产品的质量差异。
综上所述,制剂产品的开发不仅仅是产品本身的开发更重要的是方法以及标杆的开发。随着国内制药行业的大浪淘沙,制剂开发者也在越来越重视后者的开发,从起初的QC方法的直接使用到后来的多条曲线对比以及现在区分力方法的不断探索都已表明行业的不断进步。然,这条路依然很远,我们才刚刚起帆。
主要参考文献:
1.《口服药物吸收与转运》
2. Biorelevant Dissolution MediaSimulating the Proximal HumanGastrointestinal Tract: An Update
3. Application of biorelevant dissolution tests to the prediction of in vivo performance of diclofenac sodium from an oral modified-release pellet dosage form
4.溶出度实验结合计算机模拟技术评价国产β-内酰胺类抗生素的生物等效性

来源:零下274℃ 药事纵横


