您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-10-12 21:15
为配套新修订的《医疗器械监督管理条例》和《医疗器械注册管理办法》,国家药品监督局于2021年9月30号发布了《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告(2021年第121号)》,对医疗器械申报资料的具体要求作出详细解释。该文件将在2022年1月1号开始实施。
新发布的医疗器械注册申报资料从形式上与原43号令相比有了较大的变化,由原来的12大类资料修改为6类,对比关系详见下图。但从资料的内容上来说变化不大。
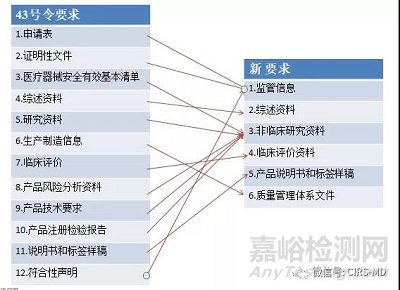
01监管信息
监管信息包含了原申请表、证明性文件和符合性声明,此外还增加了产品列表、主文档授权函、与监管机构的沟通历史记录。产品列表与原4.3规格型号的要求基本一致,需要说明拟申报产品的型号、规格、结构组成、标识、尺寸等信息。而主文档授权函则是为了适应主文档登记制度的设立,一定程度上也解决了器械申请人无法提供原材料核心信息的问题。与监管机构的沟通记录指的是通过会议形式的沟通或者有既往申报历史的可根据情况提交相关沟通记录。
02综述资料
与43号令相比,新的综述资料的要求增加了申报产品上市历史的要求,适用时,申请人应提供拟申报产品在不同国家(地区)的上市情况、不良事件和召回情况、销售情况及不良事件及召回率。
03非临床研究资料
非临床研究资料包括了原风险管理资料、技术要求及检验报告、医疗器械安全和性能基本原则清单、研究资料、稳定性研究资料、免临床评价产品的对比资料。此外,增加了非临床文献资料,若申报申报产品具有非临床文献应提供文献目录及原文。研究资料中亦需关注化学/材料表征、互操作性。医疗器械安全和性能基本原则清单的内容也进行了调整。
04临床评价资料
临床评价资料部分应阐述产品的研发背景包括现有的诊断或治疗方法涉及的器械的临床应用情况,申报产品与现有诊断和治疗方法的关系、预期达到的临床效果;临床评价涵盖范围;临床评价路径;不同路径的临床评价具体资料。
05说明书和标签样稿
说明书与标签的要求与原43号文是一致的,并无变化。
06质量管理体系文件
新的资料要求变化最大的就是需要在注册申报时提交质量管理体系文件。质量管理体系文件除了原先注册时提交的生产制造信息外,还包括原体系核查申请资料以及体系内的程序文件。这意味着,企业必须完成质量体系建设,做好体考准备后才能提出产品注册申请。
延续注册资料内容上与原要求一致,变更注册文件增加了适用时质量管理体系文件的要求,适用情况包括变更的具体原因或目的涉及产品设计、原材料、生产工艺、适用范围、使用方法变化的,需要针对变化部分进行质量管理体系核查的情形。

来源:CIRS医械合规动态


