您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-04-12 13:41
引用本文
魏澜,聂涛,王爱君*.2015~2019 年医疗器械生产企业飞行检查情况综述[J].中国食品药品监管.2021.02(205):54-64.
2015~2019 年医疗器械生产企业飞行检查情况综述
Overview of Unannounced Inspections of Medical
Device Manufacturers from 2015 to 2019
魏澜
国家药品监督管理局食品药品审核查验中心
WEI Lan
Center for Food and Drug Inspection of NMPA
聂涛
国家药品监督管理局食品药品审核查验中心
NIE Tao
Center for Food and Drug Inspection of NMPA
王爱君
国家药品监督管理局食品药品审核查验中心
WANG Ai-jun
Center for Food and Drug Inspection of NMPA
摘 要
Abstract
为加强医疗器械监督检查,强化医疗器械安全风险防控, 2015 年6 月原国家食品药品监督管理总局发布了《药品医疗器械飞行检查办法》。同年9 月原国家食品药品监督管理总局医疗器械监督管理司聚焦高风险产品,突出重大有因事项,启动了医疗器械生产企业的飞行检查工作。本文通过对2015~2019 年医疗器械生产企业飞行检查的数量、品种类型、检查、结果以及发现的缺陷问题进行全面地汇总、统计、分析,提出了医疗器械生产企业飞行检查的工作建议。
In order to strengthen the supervision and inspection of medical devices, and reinforce risk control for medical devices, the former China Food and Drug Administration (CFDA) issued the Provisions for Unannounced Inspection of Drugs and Medical Devices in June 2015. Starting from September of 2015, CFDA’s Department of Medical Device Regulation launched unannounced inspections of medical device manufacturers by focusing on high-risk products and major causes for concern. This article summarizes and analyzes the number of unannounced inspections conducted on medical device manufacturers, types of products inspected, inspection results and problems exposed during inspections from 2015 to 2019, and puts forward suggestions on unannounced inspections of medical device manufacturers.
关键词
Key words
医疗器械;飞行检查;综合分析;存在问题;建议
medical devices; unannounced inspections; comprehensive analysis; existing problems; suggestion
医疗器械飞行检查是医疗器械监管部门围绕安全风险防控开展的不预先告知的监督检查,具有突击性、独立性、高效性等特点。《医疗器械生产质量管理规范》[1](以下简称《规范》)是医疗器械生产企业确保产品质量的基本准则,也是企业的第一责任。飞行检查有效地实现第一时间第一现场,在调查问题、强化监管、震慑违法等方面发挥了重要作用。
国家药品监督管理局医疗器械监督管理司按照《药品医疗器械飞行检查办法》要求[2], 坚持以问题为导向,以风险管控为核心,以“双随机、一公开”为基本原则,即随机抽取企业和检查员,公开检查结果。持续对医疗器械生产企业实施飞行检查,对检查发现问题及处理措施进行公告。不仅对企业形成强大威慑力、强化了企业自觉实施质量管理体系的意识,而且也提升了检查员善于发现问题、依法依规处理问题的能力。医疗器械生产企业飞行检查的数据分析,为医疗器械生产监管风险防控决策提供了重要依据。
一、总体情况
按照职责分工,国家药品监督管理局医疗器械监督管理司根据监管重点制定年度医疗器械生产企业飞行检查计划, 国家药品监督管理局食品药品审核查验中心负责检查任务实施。2015~2019 年期间,以问题为导向,聚焦高风险三类医疗器械产品,共选派了国家医疗器械检查员507 人次, 对327 家医疗器械生产企业实施飞行检查(图1)。
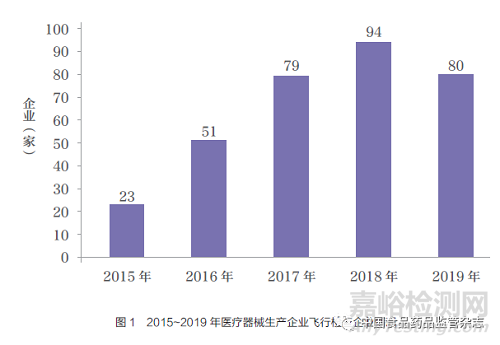
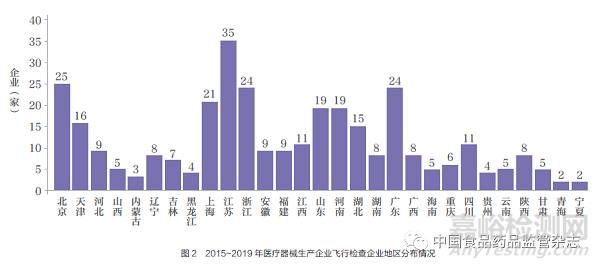
众所周知, 医疗器械生产企业主要集中在北上广和长三角地区。为全面了解医疗器械生产企业的情况,在按比例随机抽取企业的原则基础上,兼顾省份的覆盖,检查的覆盖面逐年扩大,由2015年9 省(自治区、直辖市)医疗器械生产企业飞行检查,扩大到2019 年除新疆、西藏外的29 省(自治区、直辖市)。其中,排名前5 位的是江苏省35 家、北京市25 家、广东省24 家、浙江省24 家、上海市21 家(图2)。
(一)检查品种
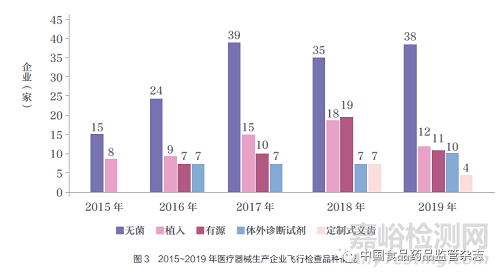
随着飞行检查的企业数量逐年增加,检查的品种范围也不断扩大,2015 年飞行检查仅涉及无菌类和植入类产品,且这两类高风险产品持续保持较高的检查比例,占全年检查总数量的60%以上。自2016 年起,飞行检查覆盖有源、体外诊断试剂类产品。2016 年12 月21 日,《医疗器械生产质量管理规范附录定制式义齿》[3] 发布,为实地调查企业实施情况,2018 年启动了定制式义齿产品的飞行检查。
2015~2019 年飞行检查的企业中,无菌医疗器械生产企业151 家,植入医疗器械生产企业62 家,有源医疗器械生产企业47 家,体外诊断试剂医疗器械生产企业31 家,定制式义齿11 家,其他25 家(图3)。
(二)检查原由
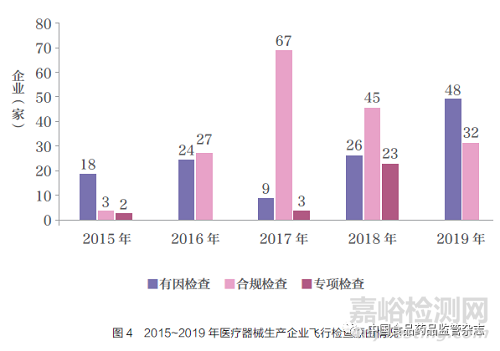
《药品医疗器械飞行检查办法》第八条规定,开展药品医疗器械飞行检查的情形主要有投诉举报或者其他来源的线索表明可能存在质量安全风险的、检验发现存在质量安全风险的、药品不良反应或者医疗器械不良事件监测提示可能存在质量安全风险的、对申报资料真实性有疑问的、涉嫌严重违反质量管理规范要求的、企业有严重不守信记录的、其他需要开展飞行检查的情形。
随着监管需要的不断提升,现阶段飞行检查主要包括合规检查,因投诉举报、检验、不良反应监测等风险信号启动的有因检查,以及某类产品的专项检查。2015~2019 年共开展有因检查125 家,占比38.2% ;合规检查174 家,占比53.2% ;专项检查28 家,占比8.6%(图4)。
(三)检查结果
医疗器械生产企业飞行检查评定原则规定[4],现场检查发现企业存在严重缺陷的,或不符合项可能对产品质量产生直接影响的,企业必须停产整改;如仅存在一般项目不符合要求,且不对产品质量产生直接影响的,企业必须限期整改。
国家药品监督管理局网站定期公开飞行检查结果及其缺陷情况,对于责令停产整改的企业予以通告,限期整改的企业予以公告。检查结果公开一方面震慑被检查的企业,提升企业对质量体系的敬畏和执行规范的自觉性;另一方面,其他企业也可以间接学习研究缺陷问题自查自纠,促进《规范》的有效实施。
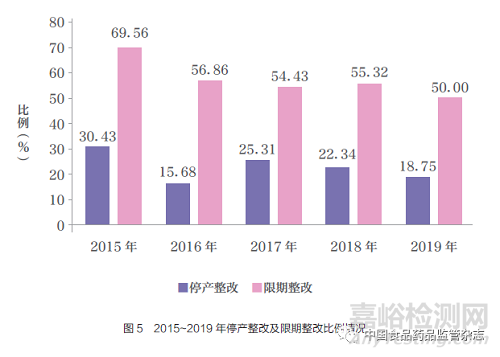
总体来看,无菌类和植入类产品飞行检查停产整改比例较高。与2015 年和2017 年相比,2016 年对无菌类和植入类产品检查比例相对较小,故停产整改数量偏低。随着飞行检查进程的推进,企业质量管理水平不断提升,检查尺度一致性持续提高, 从2017 年开始飞行检查结果为停产整改的企业数量逐年下降,2019年已不足20%(图5)。
二、现场发现的主要问题
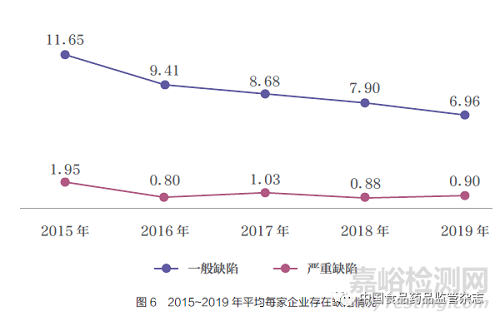
2015~2019 年,医疗器械生产企业飞行检查,现场发现缺陷3056 项,其中严重缺陷323 项,一般缺陷2733 项。平均每家企业发现一般缺陷数量逐年降低,严重缺陷数量基本持平(图6)。
(一)无菌医疗器械生产企业
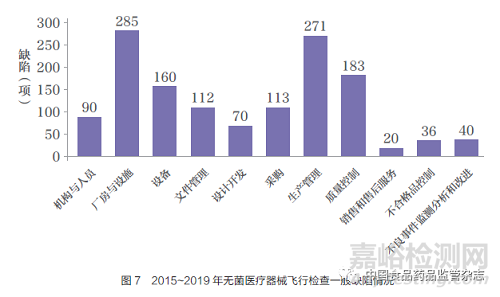
2015~2019 年对151 家无菌医疗器械生产企业实施飞行检查, 共发现一般缺陷1380 项,严重缺陷173 项。通过对缺陷项目统计分析发现,严重缺陷主要集中在生产管理和质量控制,分别是46 项(占比26.6%)和44项(占比25.4%)。一般缺陷主要集中在厂房与设施(285 项,占比20.7%)、生产管理(271 项,占比19.6%)、质量控制(183 项,占比13.3%) 和设备(160 项,占比11.6%)(图7)。
2015~2019 年无菌医疗器械生产企业飞行检查中,缺陷问题主要表现如下。
厂房与设施:物料无标识、物料混放,货位卡记录信息不全;厂区路面不平整,杂草丛生;未对洁净区的压差进行有效控制;厂房缺少适当的照明、温湿度和通风控制条件。
生产管理:生产记录缺失或记录不全,记录与规定不符,生产记录不能满足追溯要求;未对中间品进行标识;未对灭菌柜过载时灭菌程序的适用性进行评估;验证报告中未对物料的装载方式及最大装载数量进行确认;灭菌参数记录与实际不符;灭菌柜设备操作规程中无灭菌货物装载图和生物指示剂布点图;未对进入洁净区的产品进行净化处理;未对净化程序的有效性进行验证。
质量控制:未对设备进行校准和标识;未能提供校准或检定记录;设备使用环境不满足规定要求;未能提供原始检验记录;检查规程中的技术参数指标与技术要求中规定的不一致;未配备技术要求中关键项目的检验设备;未按照规定要求对产品进行抽样;检验记录无法追溯;检验记录内容不全;检验记录与实际操作不一致;未按照产品和工艺特点制定留样管理规定;留样量不足以满足全性能测试要求;留样样品实际数量与留样记录不一致;未对留样室环境进行监测;不能提供水质检测报告或记录;未按照规定的检测频次进行检测。
(二)植入医疗器械生产企业
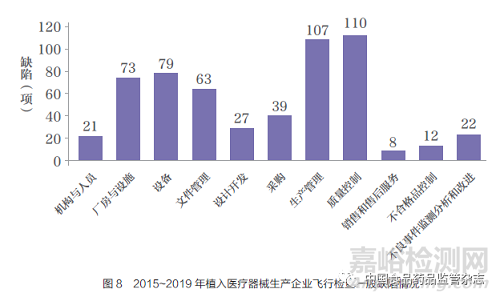
2015~2019 年对62 家植入医疗器械生产企业实施飞行检查,现场发现一般缺陷561 项,严重缺陷58 项。严重缺陷主要集中在生产管理、质量控制和采购三个方面。一般缺陷以质量控制和生产管理方面的问题更为集中,分别为110 项( 占比19.6%) 和107 项( 占比19.1%), 其次是设备、厂房与设施方面的缺陷项,分别为79 项(占比14.1%)、73项(占比13.0%)(图8)。
2015~2019 年植入医疗器械生产企业飞行检查中,缺陷问题主要表现如下。
生产管理:生产记录缺失或记录不全,未记录部分主要设备的名称和编号,未按照规程的要求进行记录;未制定人员防护规程,未对生产过程中产生的粉尘和有害物质进行控制;环氧乙烷解析库无解析条件要求、未配备温度调节和监测设备。
质量控制:检验未按照技术要求中规定的环境下操作;尺寸过程检验缺少原始记录;产品储存期限到期后未按规定进行检测;未对产品的储存期限和储存条件进行规定;阳性菌种未标明传代次数。
采购:未按照规定对供应商进行现场审核,质量管理部门人员未参与供方评价;未能提供供方的检验报告及验收准则;原材料的质量标准中缺少国家标准中规定的项目。
(三)体外诊断试剂医疗器械生产企业
2015 年飞行检查未覆盖体外诊断试剂产品,2016~2019 年对31 家体外诊断试剂医疗器械生产企业实施飞行检查,现场发现一般缺陷262 项,严重缺陷14 项。一般缺陷项主要集中在设备、生产管理、厂房与设施等方面,分别为57 项( 占比21.8%)、40 项( 占比15.3%) 和35 项( 占比13.4%)(图9)。
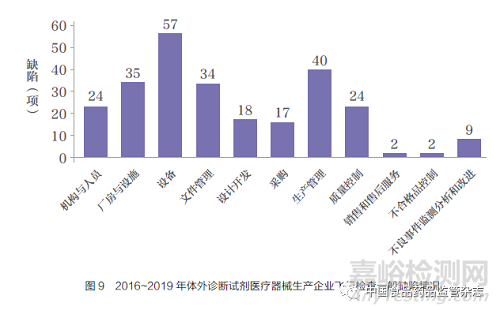
2016~2019 年体外诊断试剂医疗器械生产企业飞行检查中,缺陷问题主要表现如下。
厂房与设施:原材料库存数量与货位卡登记数量不一致;-80 ℃、-20℃ 冰箱无货位卡;未明确生物安全柜过滤器维护、保养周期及具体要求,未对阳性对照间的生物安全柜的过滤器性能进行定期检查,未对生物安全柜进行局部100 级的检测和监测;阴性血清参考品制备未在10 000级洁净级别、相对负压的区域操作。
文件管理:记录与规定不符,记录与实际不符,应记未记、错记、记录不清晰、不完整;记录涂改或销毁不规范;未能提供文件变更和分发记录;文件无作废标识;现场提供的技术文件未标识版本号,无法识别文件更改和修订状态。
生产管理:生产记录中的参数与技术要求中的不一致;记录内容不全;引物探针溶液没有批号标识;未记录设备使用信息;未明确中间品的储存期限和条件;未确认抗体反复冻融次数对其活性的影响,也未对抗体活性进行检测。
质量控制:未明确规定检验仪器不符合要求时对以往检验结果进行评价的方法;对关键检验设备的防护未做规定;来料功能性检验记录中未记录质控品的批号;菌种传代记录不完整;企业参考品配制记录中的配制过程未体现稀释倍数;参考品换代未做连续性标定相关工作;未做阴性参考品N3-N9 的病原体确认;未按照要求对参考品复验;阳性参考品无分装记录。
(四)有源医疗器械生产企业
2015 年飞行检查未覆盖有源产品,2016~2019 年对47 家有源医疗器械生产企业实施飞行检查,现场发现284 项缺陷。其中严重缺陷34 项,主要集中在质量控制和生产管理方面,分别为13 项( 占比38.2%) 和7 项( 占比20.6%)。一般缺陷250项,主要集中在文件管理、生产管理和质量控制方面,分别为39项(占比15.6%)、32 项(占比12.8%)和30 项(占比12.0%)(图10)。
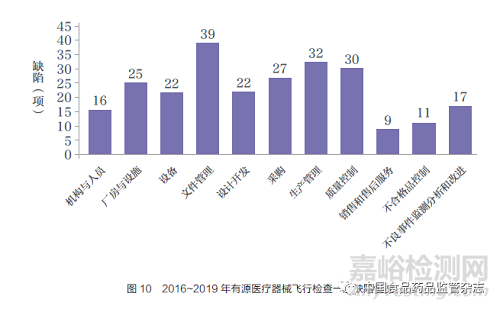
2016~2019 年有源医疗器械生产企业飞行检查中,缺陷问题主要表现如下。
文件管理:文件无编号和受控标识;文件无作废标识,作废文件被误用;文件保存时限设置不合理,原始记录不规范。
生产管理:未对特殊过程焊接进行确认和再确认;批记录中不能追溯关键元器件的序列号;生产记录不全,未记录主要设备信息;焊接工位作业区域地面标识了防静电警示线,但未铺设绝缘地垫;半导体模块没有静电防护袋;多种元器件(变压器、电容等)未采取防护措施,表面存在积尘。
质量控制:企业进货检验记录内容不全,进货检验记录中缺少对主要原材料安全性能指标的检验结果;企业不能提供抽样比例的设定依据;检验设备未按照要求定期校准。
三、思考与建议
(一)加强《规范》培训,提升企业质量体系管理水平
医疗器械生产企业是医疗器械产品质量的责任主体,也是医疗器械生产质量的第一责任人,企业自觉按照《规范》及其相关法规、产品技术要求生产是职业准则。从2015~2019 年数据分析显示,虽然平均每家企业发现一般缺陷数量逐年降低,但严重缺陷数量基本持平。主要原因是一些企业对质量体系条款的认知存在局限性,理解不到位,对于严重问题不能及时认知。目前医疗器械监管部门对企业的培训仍显不足。医疗器械监管部门需要深入研究落实“放管服”改革,在继续加大飞行检查力度的同时,指导企业走出质量体系的误区,提升企业的认知水平,使质量体系真正落实到位,确保医疗器械产品的质量安全。
(二)整合监管信息,实现智慧监管
随着社会化进程的加快,医疗器械生产企业和产品数量逐年激增,科学监管、智慧监管成为医疗器械监管进入数据时代的新课题,改进工作机制,借助智能手段,建立新的监管模式势在必行。建议对已有医疗器械生产企业监管数据库进行整合,实现及时风险预警,实时数据上报,实时分析监控。同时兼顾质量监督抽检、不良事件监测、投诉举报办理、产品主动召回以及舆情监测等信息,通过大数据技术,综合评价企业风险级别,形成高风险企业动态清单,使监管更加科学有效。
(三)尽快出台《检查员管理办法》,推进职业化检查员队伍建设
飞行检查任务的落实需要组织检查员,检查员的素质直接影响检查的工作质量,继而影响监管的决策。自2015 年原国家食品药品监督管理总局每年对检查员进行入职培训和继续教育培训,强化专业实训、统一检查尺度,以适应医疗器械飞行检查的需求。截至2020 年,已获得国家检查员证书的在岗检查员207 人,但这些检查员基本为兼职,由于检查员职业的专业性和实时性,建立职业化检查员队伍迫在眉睫。建议有关部门按照《国务院办公厅关于建立职业化专业化药品检查员队伍的意见》[5] 的要求, 尽快出台《检查员管理办法》,打造一支政治过硬、素质优良、业务精湛、廉洁高效的检查员队伍, 提升医疗器械安全风险防控能力和支撑保障水平,以满足医疗器械监管工作需要。
第一作者简介
魏澜,国家药品监督管理局食品药品审核查验中心,药师。专业方向:医疗器械质量管理体系检查
通讯作者简介
王爱君,国家药品监督管理局食品药品审核查验中心,主任药师。专业方向:医疗器械质量管理体系检查

来源:中国食品药品监管杂志


