您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-02-26 08:59
为提升我国制药行业整体水平,保障药品安全性和有效性,促进医药产业升级和结构调整,增强国际竞争能力,国家陆续出台相关文件,对提高仿制药质量、开展并加快仿制药质量一致性评价等工作提出要求。
仿制药的研发及审评应基于仿制药质量和疗效与原研药品的一致原则,其中生物等效性评价是口服固体制剂研发及审评的重要关注点。
原国家食品药品监督管理总局发布的《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》,阐述了以药动学参数为终点评价指标的化学药物仿制药人体生物等效性试验的一般原则,适用于体内药物浓度能够准确测定并可用于生物等效性评价的口服及部分非口服给药制剂,具有重要参考意义。
考虑到临床试验资源及试验费用问题,如何获得仿制药品种的生物等效性试验的豁免(如基于BCS分类的生物豁免,或者对于多规格制剂规格之间的生物豁免),越来越受到国内制药企业的关注,目前在审评实际工作中也遇到越来越多案例。
对于基于BCS分类的生物豁免,欧美监管机构及ICH都有相关指南发布,可以基于产品的溶解性、渗透性以及体外溶出特性对BCS分类1和3类的口服常释制剂申请豁免体内临床试验,仿制药质量和疗效一致性评价办公室也发布《人体生物等效性试验豁免指导原则》并陆续公布可豁免或简化人体生物等效性(BE)试验品种。
对于存在多个规格的仿制药品种,可能会涉及不同规格之间的生物豁免(即仅对部分规格进行体内BE研究,其他规格可申请BE豁免),考虑到这种生物豁免情形在实际研发及审评工作经常遇到,而且国内外的相关要求不明确,甚至技术要求存在不一致,给企业研发及实际审评工作(如境内外共同开发共同申报)带来困难和挑战。
基于此,笔者经调研国内外相关指南,重点从药学角度(如处方和体外溶出相似性)分析讨论多规格固体制剂规格间生物豁免的要求(包括常释及调释制剂),代表个人观点和审评体会,希望对仿制药及已上市化学药品仿制药的一致性评价工作提供帮助。
国内外技术要求汇总
FDA要求
根据美国联邦法规要求,申请人可以通过提交体外证据来证明体内生物等效,即豁免体内的BE研究,其中提到了对于存在多个规格的常释制剂规格之间生物豁免的要求,即可以在满足相同剂型、处方比例相似、体外溶出三方面的前提下允许规格之间的生物豁免,但是未展开进一步详细说明。
根据FDA发布的《以药动学参数为终点评价指标的仿制药生物等效性研究指南》,该指南中明确了对于不同剂型(口服液、常释和调释片剂/胶囊剂、混悬液、咀嚼片)开展BE试验的一般要求,其中提到,对常释制剂和调释制剂,若满足一定条件,部分规格的生物等效性试验可豁免,下面分别就常释和调释制剂两方面分别进行阐述。
首先,对于常释制剂,规格间的生物豁免需要满足的条件包括:①试验规格符合生物等效的要求。②所有规格可接受的体外溶出结果。③所有规格处方比例相似,且处方比例相似包含如下几种情况:不同规格之间原辅料比例相同(比如涉及25,50,100mg几种规格,50mg辅料的用量,应为25mg规格的2倍,应为100mg规格的一半)、高活性药物(即活性成分用量占比较低的情况,不同规格之间应总重相同或允许±10%差异,辅料种类相同,辅料用量调整来弥补不同规格活性成分用量差异)、其他合理解释(如通过剂型比例研究证明体内生物利用度等效)。
其次,对于调释制剂,规格间的生物豁免需要满足的条件包括:①所有规格处方比例相似。②所有规格释药原理相同。③各规格制剂体外溶出结果可接受。比例相似的定义与常释制剂相同,同时还明确了BE试验规格与其他规格之间应通过至少3种溶出介质(例如pH1.2,4.5和6.8)的f2因子来证明溶出行为的相似性,而常释制剂未对溶出提出细化要求。
近期,FDA更新发布了《新药临床试验及上市申请提交生物利用度研究的一般考虑-草案》,对新药开发过程中涉及多规格制剂的规格间生物豁免的情况进行了说明。笔者认为,虽然该指南适用范围是针对新药的临床试验申请和上市申请等,但是其中对于处方比例的相似性的定义及体外溶出数据的要求,在仿制药简略新药申请(ANDA)也可以借鉴。
其中,处方比例相似包含如下5种情况:不同规格之间原辅料比例相同、高活性药物(即活性成分用量占片芯或胶囊内容物的5%以内的情况,不同规格之间总重相同或允许±10%差异,采用相同的辅料种类,调整一种或多种辅料用量来保持总重一致)、双层片(每一层的所有组分都应该比例相似,才能判定为整体的处方相似,如果有任何一层不满足,则不能认为是处方比例相似)、符合SUPAC二级变更(即辅料用量在SUPAC-IR/MR二级变更范围内)、其他合理依据(不符合上述情况,如提供合理依据,经与审评部门沟通也可以认可)。
笔者将该指南与FDA2013年发布的《以药动学参数为终点评价指标的仿制药生物等效性研究指南》进行对比,发现该指南草案中对于处方比例相似的定义、溶出比较等进行了部分调整和更新,具体涉及如下4个方面:
①该指南明确了高活性药物是活性成分用量占片芯或胶囊内容物的5%以内。
②该指南增加了对于双层片处方相似的要求。
③该指南增加了辅料用量(占总重)不超出SUPAC-IR/MR二级变更范围的情形:对于常释制剂,辅料用量的二级变更如下:填充剂(±10%)、崩解剂(淀粉±6%,其他±2%)、黏合剂(±1%)、润滑剂(硬脂酸镁/钙±0.5%,其他±2%)、助流剂(滑石粉±2%,其他±0.2%)、薄膜包衣剂(±2%),这也与基于BCS分类的生物豁免指南中BCS3类药物豁免要求中可允许的辅料用量差异范围一致;对于调释制剂,非释药控制性辅料的二级变更参考常释制剂,释药控制性辅料的二级变更范围应≤10%。
④体外溶出明确了可采用一种推荐(如可用)的溶出方法进行规格间的溶出对比。
为了更好地帮助仿制药企业开展BE研究,除上述FDA发布的一般性的指导原则外,FDA还公布了特定药物的BE指导原则,针对具体药物提出具体指导意见,并不断在特定药物指南数据库“Product Specific Recommendations for Generic Drug Development”更新。
截至2019年2月,特定药物品种数目已增录至1600多个,涉及不同给药途径(口服占大部分,还包括舌下、肌注、吸入、鼻用、外用等)和剂型[片剂(口崩、咀嚼、泡腾、调释等)、胶囊剂、脂质体、乳膏、喷雾剂]。这些具体药物指南对于口服给药途径的片剂/胶囊剂(包括常释、调释)的规格之间生物豁免的要求与FDA发布的一般性指导原则基本保持一致。
此外,具体品种指南中还对溶出数据的提交要求进行说明:
对于常释制剂,一般都要求采用FDA溶出数据库的方法开展各规格自制制剂与参比制剂的溶出对比(12个样品);
对于调释制剂,除要求采用FDA溶出数据库的溶出方法开展自制制剂与参比制剂的各规格下的溶出对比(12个样品)外,还明确要求提供自制制剂与参比制剂的各规格(12个样本)在至少3种不同pH介质(如pH1.2,4.5和6.8缓冲液,部分品种也要求水介质)的溶出对比,溶出方法采用USP装置Ⅰ(100r·min-1)和/或装置Ⅱ(50r·min-1),或者转速适当增加。如需要,溶出介质可加入适量的表面活性剂。取样时间点应包括早期数据点(1,2,4h)后面每隔2h取样,直到80%以上药物释放。
EMA要求
根据EMA发布的生物等效研究指导原则,对于需要开展BE研究的多规格制剂品种,也明确了规格间生物豁免的可能性。
需要满足的条件如下:
①不同规格采用相同的生产工艺。
②不同规格的处方种类相同。
③不同规格原辅料组成定量成比例,即规格之间原辅料的用量比例相同。常释制剂的包衣层、胶囊壳、着色剂和矫味剂除外。当处方中活性成分用量低于5%(API占片芯/胶囊内容物的比例)时,不同规格之间应该辅料用量相同,或者不同规格之间通过调节填充剂用量来弥补活性成分用量差异并达到相同总重(片芯、胶囊内容物)。
④适宜的体外溶出数据。应开展BE规格与其他规格之间溶出相似性比较,介质通常应包括pH1.2,4.5,6.8,如选择其他介质,应提供依据。部分pH条件下不一定所有规格都满足漏槽条件,这样会导致不同规格之间的溶出会有差异。
这种情况下,建议进行自制与相应规格参比制剂的溶出对比,以确认这个现象是由于原料药而非处方组成导致的。另外,也可以选择采用相同剂量进行溶出比较以证明溶出相似性,如2片5mg规格与1片10mg规格进行比较。
与FDA不同的是,该EMA指南未针对常释及调释制剂的豁免要求分类进行阐述,根据EMA口服调释制剂研究指南,如果将体内研究结果外推用于支持其他规格调释制剂的豁免,应开展自制制剂其他规格与BE研究所用规格的溶出曲线比较,并判定相似性,而且在溶出曲线比较时,应结合对活性成分的理化性质及制剂的释放机理的考虑,合理设定取样时间点和频率。
EMA指南还对组合制剂和双层片的情况进行了说明。对于组合制剂产品,所有活性成分都应该满足处方比例的要求(考虑其中一个活性成分时,其他活性成分可算作辅料);对于双层片产品,可对每个单层分别进行考虑。
此外,还指出可以采用括号法的情况,即采用能代表极端情况的规格来开展BE研究,则其他规格可允许生物豁免,比如最高和最低规格,或者处方差异最大的2个规格。与FDA一样,EMA从2013年开始,也定期公布并更新特定药物的生物等效指南,对特定药物的BE试验方法进行说明。
国内法规
根据原国家食品药品监督管理总局2016年发布的《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》,若满足一定条件可允许豁免部分规格的生物等效性试验,并分常释和调释制剂2种情况进行说明。
首先,对于常释片剂和胶囊,一般采用申报的最高规格进行单次给药的空腹及餐后BE研究,若同时满足以下条件,其他规格制剂的生物等效性试验可豁免:
①试验规格制剂符合生物等效性要求。
②各规格制剂在不同pH介质中体外溶出曲线相似。
③各规格制剂的处方比例相似,对于制剂处方比例相似的要求与FDA基本一致,包括不同规格之间组成比例相似、高活性药物(原料药占制剂重量比例低)的情况。
其次,对于调释制剂,一般采用申报的最高规格进行单次给药的空腹及餐后BE研究,若以下条件全部满足,则可以认为调释制剂的其他规格与相应规格的参比制剂具有生物等效性:
①其他规格制剂的活性和非活性组分组成比例与试验规格的受试制剂相似。
②其他规格制剂的释药原理与试验规格的受试制剂相同。
③各规格制剂体外溶出试验结果相似。同时建议至少在3种不同pH溶媒(例如pH1.2,4.5和6.8)中通过f2值判断其他规格的溶出曲线与BE研究中受试制剂溶出曲线的相似性。
除上述指导原则外,原国家食品药品监督管理总局2015年发布的《普通口服固体制剂溶出度试验技术指导原则》,也对规格豁免的情况进行了简单说明:
对于多规格药品,当药物具有线性动力学的特点且不同剂量规格药品处方组成比例相似时,可对最大剂量规格的药品开展BE研究,基于充分的溶出度比较试验,可以豁免小剂量规格药品的体内研究。
处方组成比例相似性的判定可参见《已上市化学药品变更研究技术指导原则》中“变更药品处方中已有药用要求的辅料”项下的相应内容。
需要说明的是,与FDA/EMA不同,国内尚未有特定药物的生物等效性指南发布。中国食品药品检定研究院于2017年对EMA和FDA《特定药物的生物等效性指导原则》的有关内容开展了翻译工作并公布在官网,以期对我国正在开展的仿制药质量与疗效一致性评价工作起到借鉴和指导作用。
分析和讨论
通过上述汇总可见,国内外都明确了多规格仿制药规格间生物豁免的可能性,而且药学角度均重点关注处方和溶出度两方面,如若能满足处方相似及溶出相似的要求,原则上可基于特定规格的BE研究来豁免其他规格的体内研究。虽然整体要求基本保持一致,但相互之间仍然存在部分差异。
处方相似性的要求
根据表1,对于处方相似性的判定:
①FDA包含情况较为全面,包括原辅料等比例、低活性药物的考虑、参照SUPAC的允许变更范围以及其他可以解释情况等多个方面,操作较为灵活。
②国内对于处方相似性的定义在《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》有较为明确的说明,基本上沿用FDA2013年发布的《以药动学参数为终点评价指标的仿制药生物等效性研究指南》的定义,但对于后续FDA更新的关于高活性药物的定义、SUPAC-IR/MR二级变更允许范围、双层片的要求等方面未涉及,仅在《普通口服固体制剂溶出度试验技术指导原则》指出也可参考《已上市化学药品变更研究技术指导原则》中“变更药品处方中已有药用要求的辅料”项下的相应内容来评估处方比例相似。
③EMA的定义主要包括原辅料组成比例相似、高活性药物2种情况,基本与FDA要求一致,而且也对双层片处方相似的判定情况进行了说明。
体外溶出的要求
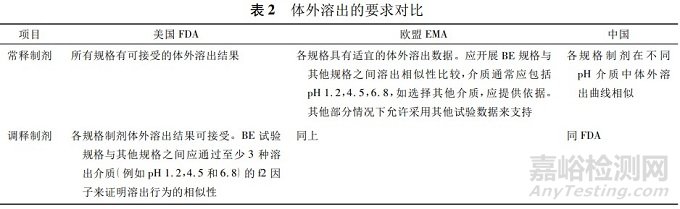
根据表2,对于常释制剂的规格间豁免:
①国内要求各规格制剂在不同pH介质中体外溶出曲线相似,未提供进一步的解释和说明。
②FDA《以药动学参数为终点评价指标的仿制药生物等效性研究指南》仅指出所有规格有可接受的体外溶出结果,阐述比较模糊,未对“可接受的体外溶出结果”进行明确说明;FDA早期发布的常释制剂的体外溶出试验指导原则中提到可基于适当的体外溶出试验来支持常释制剂规格间的BE豁免,体外溶出试验建议BE规格与其他规格之间进行溶出曲线比较,但仍然未明确溶出试验条件及要求;
笔者进一步查阅,为支持常释制剂规格之间的生物豁免,FDA认为,如果已经建立了适宜的溶出方法,而且溶出结果表明溶出特性与规格无关(不同规格溶出相似),通常可进行一种介质下的溶出曲线比较;如果尚未建立适宜的溶出方法,应进行3种介质(pH1.2,4.5,6.8)下的溶出曲线比较。
③EMA明确要求应开展自制制剂与BE规格样品之间的溶出比较,而且EMA指南中的考虑较为周全,提到部分情况下允许采用其他试验数据来支持的情形,如部分pH条件下不一定所有规格都满足漏槽条件,这样会导致不同规格之间的溶出会有差异,可进行自制与相应规格参比制剂的溶出对比,或者也可以选择采用相同剂量进行溶出比较以证明溶出相似性,整体上EMA可操作性和灵活性更强一些,而国内及FDA在指导原则中并未描述允许采用其他试验数据来支持的情况。
笔者检索到FDA文章对具有规格依赖性溶出特征(即由于规格的不同导致溶出曲线不相似)的产品也会予以特殊考虑,即规格之间满足处方相似但是不满足溶出相似性,仍然可以基于对差异的因素合理分析(如溶解度、处方等)提供依据来申请生物豁免,以避免不必要的体内研究。
根据表2,对于调释制剂规格间的豁免,国内外的要求较为明确,而且要求也基本保持一致,在处方比例相似的基础上,都要求进行自制制剂不同规格之间在多种溶出介质下的溶出对比研究。经进一步查阅FDA相关指南及FDA文章,为支持调释制剂规格之间的生物豁免,在满足规格间处方比例相似和释药机理相同的前提下,应根据调释制剂的不同类型来提供相应的溶出数据:对于小丸类缓释胶囊,不同规格之间仅是小丸数量不同,在推荐的溶出方法下各规格之间应溶出相似;对于缓释片,BE试验规格与其他规格之间至少3种溶出介质(例如pH1.2,4.5和6.8)溶出相似;对于双相释放的迟释制剂,可采用标准介质无需多种介质下进行溶出比较。
值得一提的是,即使各监管机构都发布了规格之间生物豁免的指导原则,但是考虑到调释制剂的复杂性,以及体内外相关性受到多个因素(如活性成分、产品设计、处方因素、溶出方法)相互作用,已有相关案例提示,调释制剂即使满足指南规格间生物豁免要求(如处方相似、体外溶出相似)仍然存在生物不等效的风险。
FDA也曾经发生过缓释制剂(Impax/Teva公司开发的抗抑郁产品盐酸安非他酮缓释片)基于体外数据豁免BE上市后由于疗效问题再撤市的情况。不管是常释还是调释制剂,FDA都建议申请人提供所有规格自制和参比制剂的溶出数据,用以评估相互之间的溶出差异。
EMA没有强制要求提供参比制剂的溶出数据,EMA仅要求提供自制制剂规格间的溶出比较,但EMA建议提供BE研究批次(包括受试制剂及参比制剂)在不同缓冲介质(pH1.2,4.5,6.8)以及QC介质的溶出对比。
总结
在仿制药产品研发及一致性评价研究过程中,企业应结合每个品种的剂型特点、自制制剂规格之间的溶出对比情况、与参比制剂的溶出对比情况、活性药物成分(API)溶解情况(漏槽条件)、处方相似性情况,提供充分的试验数据,同时参考国内外相关指南要求,综合评估,为规格间的生物豁免提供支持依据。同样,在审评工作中,应根据企业提供的试验数据,并结合品种和剂型特点,综合评价规格之间BE豁免的合理性。
此外,随着国内制药企业国际化步伐加快,越来越多国内仿制药企业走中、美、欧及境内外共同开发共同申报的开发策略,考虑到国内外监管机构在技术要求上仍然存在部分差异,如计划申请规格间BE豁免,企业在研发之初就应对参比制剂及规格选择的合理性、规格之间的处方相似性、溶出比较数据的科学性和全面性等多方面综合考虑,必要时与审评机构进行沟通交流,以尽可能满足不同监管机构规格间生物豁免的要求,避免不必要的重复工作。

来源:《中国新药杂志》


